

无参比产品仿制政策落地(附解读),进目录集不算过评,新机会到来



国家药监局关于无参比制剂品种仿制研究的公告(2023年第130号),2023年10月12日。
为满足患者临床用药需求,保障药品安全、有效、质量可控,鼓励申请人选择国家药品监管部门公布的参比制剂目录中收载的品种进行仿制研究。对于参比制剂目录中未收载的品种,即无参比制剂品种,经评估如有预期临床价值,申请人可开展仿制研究。现就无参比制剂品种仿制研究有关要求公告如下:
一、坚持高标准、严要求。以问题为导向,深化药品审评审批制度改革,按照为公众提供高质量的仿制药、促进仿制药产业高质量发展的原则开展相关工作。
二、坚持以临床价值为导向。所仿制的品种应符合当前科学认识和临床诊疗需求及实践,作为主流药品被广泛使用,且具备不可替代性特征,同时有足够临床试验数据支持临床获益大于风险。申请人应充分评估拟申报品种的预期临床价值,并通过良好设计的临床试验证明其预期临床价值。
三、坚持最严谨的标准,提升产品质量。申请人应基于现行技术要求开展仿制研究,并对已上市同品种药品开展全面质量评估。仿制药品质量不低于研究充分、上市基础好或在相应疾病领域市场份额较大的已上市品种。
四、申请人在充分评估无参比制剂品种的预期临床价值的基础上,如拟仿制,应向国家药监局药品审评中心(以下简称药审中心)提出沟通交流申请(Ⅲ类),提交相关研究资料。药审中心应在规定时间内组织专家和申请人对药品预期临床价值进行沟通,沟通交流结果供申请人参考,并以适当形式公开。后续相同品种如无特殊情况,可参考已公开的沟通交流结果。
五、对于经初步判断具有预期临床价值的品种,申请人完成相关研究后,按照现行临床试验申请程序提出临床试验申请,临床试验申请审评过程中将充分参考沟通交流结果。申请人完成临床研究后,参照现行仿制药注册分类提出上市申请。药审中心应严格按照现行技术要求对仿制药申请开展审评,对于质量符合要求、有充足证据支持其临床价值的,予以批准。
六、开展临床研究的无参比制剂品种仿制药批准上市后纳入《新批准上市以及通过仿制药质量和疗效一致性评价的化学药品目录集》。无参比制剂品种的已上市仿制药不属于一致性评价范畴,相关补充申请批准后不适用一致性评价有关政策。
七、本公告自发布之日起实施。此前规定与本公告不一致的,以本公告为准。
《国家药监局关于无参比制剂品种仿制研究的公告》政策解读
一、《国家药监局关于无参比制剂品种仿制研究的公告》(以下简称《公告》)起草的背景和考虑是什么?
(一)仿制药注册分类改革
2015年8月发布的《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)将仿制药由“仿已有国家标准的药品”调整为“仿与原研药品质量和疗效一致的药品”,并调整药品注册分类,要求仿制药审评审批要以原研药品作为参比制剂,确保新批准的仿制药质量和疗效与原研药品一致。对改革前已经批准上市的仿制药,按照与原研药品质量和疗效一致的原则,分期分批进行质量一致性评价。
2016年3月原食品药品监管总局发布《关于发布化学药品注册分类改革工作方案的公告》(2016年第51号),对化学药品注册分类进行了调整,新注册分类中仿制药3类、4类和5.2类技术要求一致,均为应与原研药品的质量和疗效一致。
2020年发布的《药品注册管理办法》(市场监管总局令第27号)和《化学药品注册分类及申报资料要求》(国家药监局通告2020年第44号)明确,化学药品3类、4类和5.2类仿制药均要求与参比制剂的质量和疗效一致。
(二)仿制药参比制剂制度建立
随着化学仿制药注册分类的调整,与此相关的参比制剂概念也逐渐发生变化。国发〔2015〕44号文明确“可以选择原研药品,也可以选择国际公认的同种药品”。随着改革深入,基于新注册分类,《国家药监局关于发布化学仿制药参比制剂遴选与确定程序的公告》(2019年第25号)明确,参比制剂目录原则上应收载原研品种。《化学药品注册分类及申报资料要求》(国家药监局通告2020年第44号)明确,仿制药是按照原研药品是否在境内外上市予以区分,并要求与参比制剂质量和疗效一致。
(三)仿制药注册分类改革成果
经过药品审评审批制度改革,我国已建立与国际接轨的化学仿制药注册制度与技术评价体系,即化学仿制药应与原研药品质量和疗效一致;原研药品是指境内外首个获准上市,且具有完整和充分的安全性、有效性数据作为上市依据的药品;参比制剂目录原则上应收载原研品种,仿制药申请人应选择国家药品监管部门发布的参比制剂目录中收载的品种进行仿制研究。我国现行化学仿制药注册制度核心是以原研药品为标杆,为公众提供高质量仿制药。
(四)无参比制剂品种形成原因
在实施化学药品注册分类改革前,我国既往的仿制药研发是“仿已有国家标准的药品”,未要求以原研药品作为对照进行仿制研究。在2002年、2005年、2007年发布的《药品注册管理办法》中均规定,仿制药是指仿制已批准上市的已有国家药品标准的原料药或者制剂。按此分类的化学仿制药,一部分是对原研药品仿制的品种,但未与原研药品进行质量和疗效一致性的评价;另一部分是对非原研药品仿制的品种。前者可以按照化学仿制药注册分类及技术要求,选择参比制剂目录中的原研药品开展质量和疗效一致性研究。而后者上市时间早,缺少完整充分的安全性有效性证据、产品质量低、缺乏循证医学证据,特别是临床价值存疑,既无法实现与原研药品开展质量和疗效一致性评价,也无法为其找到仿制标杆。这就是无参比制剂品种形成的原因,也是我国仿制药发展中出现的独有历史遗留问题。
无参比制剂品种,一方面,由来已久,情况复杂,有些确有临床需求,例如葡萄糖、氯化钠注射液、维生素等;另一方面,随着新技术的发展和医学诊疗水平的进步,有的品种面临被替代或淘汰,存在是否有必要仿制的问题。总的来讲,这类药品首先应解决是否具有临床价值问题,并通过获得完整充分的安全性有效性数据,按现行要求全面提升质量申报上市。如简单地批准此类药品上市,不仅对现有的以原研药品为参照的仿制药注册制度产生冲击,对我国仿制药的高质量发展产生负面影响,也不能满足临床患者对高质量仿制药的客观需求。
为了巩固深化我国药品审评审批制度改革成果,在保证化学药品仿制药高质量发展前提下,妥善解决无参比制剂品种目前没有仿制申报路径和相关技术要求的问题,基于这类品种的特点,对于无参比制剂品种,坚持高标准、严要求。
二、《公告》的适用范围是什么?
首先,《公告》中的无参比制剂品种均为化学药品;
其次,企业对无参比制剂品种开展仿制研究时,在研发立项阶段就应对已上市同品种进行全面调研,判断是否存在具有完整和充分的安全性、有效性数据作为上市依据的原研药品,并按程序申请遴选参比制剂,如参比制剂目录中未收载,即为无参比制剂品种。
三、无参比制剂品种的临床价值如何进行评估?
无参比制剂品种一般上市时间早,缺少完整充分的安全性有效性数据,缺乏循证医学证据,特别是临床价值存疑,存在是否有必要仿制的问题。所以,这类药品首先应解决是否具有临床价值问题,也就是通过临床试验获得完整充分的安全性有效性数据,证明其临床价值。境内外已有公开数据,以及之前公示的《临床价值明确,无法推荐参比制剂的化学药品目录》中所说的“临床价值明确”,不能作为自证临床价值的证据。基于无参比制剂品种的特点,必须坚持高标准、严要求,通过临床试验确证该品种的临床价值。
四、药学研究申报资料和技术要求中 “提供不同来源已上市同品种”、“多批”等概念分别指什么?“质量研究资料”的来源是哪里?
“提供不同来源已上市同品种”是指该品种来源于不同上市许可持有人,“已上市”同品种的范围是指在全球上市的药品;“多批”具体指需要几个批次的产品,可按照现行仿制药研发要求确定。“质量研究资料”是指申请人自行开展相关研究的研究资料。申请人承担主体责任,对拟开发产品进行充分的质量研究。
五、无参比制剂品种申报仿制的注册分类是什么?
《化学药品注册分类及申报资料要求》(国家药监局通告2020年第44号)中已明确现有化学仿制药分类分为3类(仿制境外上市境内未上市原研药品)、4类(仿制境内已上市原研药品)、5.2类(境外上市的仿制药申请在境内上市)。虽然无参比制剂品种不存在原研药品概念,但是可按照境内境外是否已有该产品上市划分,参照现有仿制药注册分类申报。
六、对于不发布临床价值明确的无参比制剂品种目录和可不进行临床试验品种目录的考虑是什么?《无参比制剂品种开展仿制研究的技术要求和申报资料要求(试行)》中列举的可不再进行临床研究的品种能否不进行沟通交流,直接提出上市申请?
无参比制剂品种已上市多年,很多已被后续品种替代,如发布目录,容易使业界产生国家鼓励此类产品研发和申报的误解。另外,制订此类品种目录可操作性差,临床价值需多部门、多专业进行专家论证,短时间内无法满足申请人各种各样的研发立项需求。
对于具体品种而言,因适应症特点、安全有效性特征、剂型、给药途径、制剂复杂性等均有所不同,能否豁免临床试验受多因素影响,无法发布清单把所有品种逐一列举出来。需要企业在充分论证的基础上进行讨论研究和判断。即使《无参比制剂品种开展仿制研究的技术要求和申报资料要求(试行)》中列举的某些基础输液用产品可以不进行临床试验,此类产品也应具体品种具体分析,不能一概而论。因此,已列举的品种也需进行沟通交流,进行预期临床价值评估。
七、对于已受理无参比制剂品种的处理原则
按照坚持公开、公平、公正的原则,对于已受理的品种严格按照《公告》要求开展工作,统一标准,集中处理。对于按照一致性评价申请受理的无参比制剂品种,将按照普通补充申请进行审评,审评审批通过的,不给予“通过一致性评价”的结论。对于仿制药上市申请,也将按照《公告》中对临床价值评估、药学研究、临床研究的有关技术要求开展审评。
在医保目录无参比制剂的(正式公布为准)品种目前有816个(按着集采通用名统计),明细如下;

(信息来源:NMPA CDE 国家医保局 风云药谈整理)
大品种也有不少,举例如下;
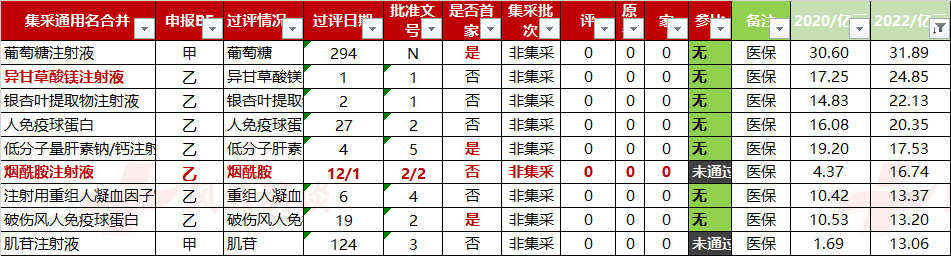
(信息来源:NMPA CDE 国家医保局 风云药谈整理)
问题1、基础输液与营养药不再进行临床研究。
这个意思很清晰,就是这两类不需要做大临床、不用做BE给批准。
文件还特意举例维生素B6,维生素B6片都集采了,维生素B6注射液首家杭州民生都快要过评了,两个产品也都有参比制剂。
维生素B6注射液的适应症也不仅仅是营养补充,适应症还比较多,联合止吐等等。
是不是只要是属于基础输液、营养药(适应症沾边)就全部不用?
对于这类产品其实挺多的,主要集中在贝朗、费卡等几个企业的产品,同时还包括维生素类、肠内营养等。
相对于维生素类产品,肠内营养产品的获批更加有难度,这个问题在CDE这边也存在争议,这类产品在国外主要以食品属性存在。
问题2、申报与临床设计
4.1 临床价值评估 申请人应首先评估无参比制剂品种的临床价值。
①适应症定位明确,给药方案具体清晰。
②具备不可替代性特征,临床有有效。
③我国人群中开展临床试验的获益风险评估。
4.2 临床试验要求
①临床试验方案设计应按照相关适应症的临床试验技术。
②应根据药物适应症特点合理选择对照药物。
③经评估, 认可该品种具有预期临床价值且临床试验方案设计科学合 理的,同意开展临床试验。
这一点就卡的比较严苛,这也就决定了批文多的产品不太可能会有企业去做大临床的。
这类产品比较大的,比如银杏叶提取物注射液、烟酰胺注射液、磷酸奥司他韦颗粒、人纤维蛋白原、注射用尿激酶、谷胱甘肽片、多烯磷脂酰胆碱口服常释剂型、盐酸克林霉素棕榈酸酯分散片、氢溴酸山莨菪碱注射液、盐酸达克罗宁胶浆、水飞蓟宾胶囊。
等等吧,这类型的产品最多,单品销售规模一般也能在5亿以上。
就算是你想做,首先要取得临床试验获批,其次,要在试验设计的时候设计的非常巧妙,这需要强大的专业能力+社会学能力。
问题3、有价值的产品不多
从政策角度讲,很多企业都关注这个政策,在医保目录内的产品有价值的实际上不是很多。

(信息来源:国家医保局 药融云 米内等 风云药谈整理)
对于在医保目录内的身份的产品是很有利的,遴选不到参比起码可以做一次质量提升。
问题的关键是,批文有几家,没有什么市场,还需要话大几千万做临床批出来一个批文,类中鸡肋型的产品商业价值在哪里?
如果说,花几千万做一个没有什么商业价值的产品,那直接买这个批文好不好呢?不可能有批文的企业审批资料都丢了吧。
问题4、国外上市有价值的产品挖掘
这个问题值得延伸去展开,国外以上市产品没有参比地位,用FDA的标准就是即不在RLD也不在RS里。
但是呢,这个产品在中国市场还比较有价值,给大家举几个例子,地佐辛注射液、氟比洛芬注射液等不少中国上市的产品都有这类情况。
大体上就是欧美日主流国内参比制剂认可的国家绝大部分没上市或者退市了,在国内销售的还不错的情况。
不过,在欧美日等国家上市不具有RLD也不在RS等参比地位的产品,在临床设计的时候怎么办办呢?
这种情况估计临床设计都帮不上什么大忙,社会学更加关键。
结合问题3与问题4,就是国内的锚定规模大的做,国外的锚定中国市场特色定(每个企业、个人理解不一样),只要在你的逻辑认知里觉得有商业化价值。
那---就是值得做的产品。
这里要注意一个问题,就是国外产品不具有参比地位,然后呢按着新的注册分类去申请,产品批下来的数据在申报前还是需要确定的。
以美国获批未在中国上市的产品(包含通用名相同剂型不同的产品)为例,有将近1500个产品还未在国内上市。
当然,各种情况都有,不一定都是没有参比的。
数据量太大还要翻译,哪位大神梳理好分享一下?

(信息来源:FDA 风云药谈整理)
主要关注的就是无申报的919个产品吧,还有其他阶段的一般进度也做不过大企业。
问题3、问题4就是解决了一大部分无参比不可仿制的情况。
从个人的经验判断,在这一轮的无参比仿制产品申报的竞赛中,集采专业户的一些企业应该也不会落下的。
有所不同的是,估计传统的集采大户扬某某、恒某某、天某某不太会凑这个热闹。
但是对于仿制药过评按通用名算有十个以上的企业,这个政策是有着莫名的吸引力。
有几个观点比较困惑,希望各位解惑。
疑惑1,无参比仿制药的批文会因为这个政策,大量的被淘汰?--这个逻辑的严谨政策支撑点在哪里?
疑惑2,无参比的赛道打开,过评格局发生巨大变化,集采将迎来重大影响---请问这个怎么重大影响?且不说集采目前需要五家,就是临床设计这关有多少企业能过,那些大品种有原控的你也拿不到原料,做临床也不是糖豆倒一下就出来一大堆。
这个政策来了,看申报、获批肯定是难的,但是,再难不也是出现的新的政策机会点么?
不感兴趣
看过了
取消

 打赏
打赏
不感兴趣
看过了
取消
精彩评论
相关阅读




中国医院排行榜















 010-82736610
010-82736610
 股票代码: 872612
股票代码: 872612



©2012-2023 北京华媒康讯信息技术股份有限公司 All Rights Reserved. 注册地址:北京 联系电话:82736610
 京公网安备 11010802020745号
京公网安备 11010802020745号


