
共同进步发展
药苑杂谈
医药人的乐园,分享探讨药学知识,
共同进步发展

1.药品杂质分析

2.基因毒性杂质的分类、限度及控制策略
潜在基因毒性杂质的分类


(基于致癌性和诱变性)-ICH M7
基因毒性杂质的判定与风险评估

基因毒性杂质的限度

TD50线性外推法:
TD50值:半数致癌量( mg/kg/day)
线性外推原理:将患癌风险由1:2线性外推至1:100,000
线性外推公式:

TD50数据来源
CPDB(carcinogenic potency database,http://toxnet.nlm.nih.gov/cpdb/cpdb.html)
里面有1547种致癌物质的列表,CAS号,TD50数据。
PDE法
计算公式:

来源:ICHQ3C
TTC法
Concept:一个具有诱变性的杂质每天每人摄入1.5μg时其风险被认为是可以忽略的(终生暴露情况下理论的患癌风险小于十万分之一)
TTC=1.5μg/day

TTC是一个风险管理工具,采用的是概率的方法,不能理解为完全无风险的保障。
TTC(1.5μg/day)是通过对已有致癌性数据的化合物可摄入量进行综合评价得出的值(TD50线性外推),所以TTC值也被认为是保守的。
LTL法
Concept:假定可接受的累积终生剂量(1.5μg/天X25,550天=38.3mg)在终生摄入期间是均匀分布在这些天数中的,这样诱变性杂质的日摄入量可以高于平均终生日暴露量,而其风险水平仍与每日或非每日治疗方案相持平(Haber 规则)。
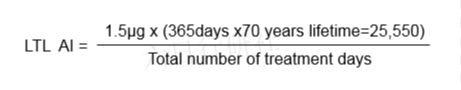
对于间歇性给药:


来源:ICHM7
总结
LE法:适用于具有致癌性数据的诱变杂质,即分类表格中的Class 1类杂质,或化学结构与已知致癌物类似的诱变性杂质(没有致癌性数据),也可以采用此方法来计算可接受摄入量。
PDE法:适用于具有真实阈值证据(NOEL/LOEL)的诱变杂质,对于有阈值的Class 1类杂质,或比TD50外推法准确。
TTC法:用于长期治疗用药物中的诱变杂质(>10年),且无致癌数据与阈值证据,即分类表格中的Class 2与Class 3类杂质,TTC法也被认为是保守的。
LTL法:TTC法的细化升级版(引入暴露时间的概念),适用杂质类型与TTC法一致。
基因毒性杂质的控制策略
方法1:在原料药中建立合适的分析方法进行控制,按照GTI/PGIs可接受 限度(e.g. 1.5μg/day)设定杂质的质量标准。
对于在较后合成步骤中引入的杂质,一般采用此方法,如:在最后一步引入GTI/PGIs。
当满足如下情况时,可以采用定期检测(periodic verification testing/skip testing): 原料药中的诱变杂质在至少6个连续的中试批次或3个连续的生产批次中,测得结果均低于限度的30% 。
注意:没有提到不订入质量标准的方式。
方法2:在原料(非原料药)、起始物料/中间体中建立合适的分析方法进行控制,按照GTI/PGIs可接受限度设定杂质的质量标准。
CDE推荐控制策略:中间体控制+终产品控制。
方法3:在起始物料、中间体中建立合适的分析方法进行控制,但是根据化学知识以及后续工艺对杂质的清除能力,将GTI/PGIs的质量标准设定为高于原料药中的可接受限度,同时保证按照此限度控制,原料药中的该杂质水平低于可接受限度(通常要低于限度的30%)。
采用此法时要有实验数据的支持(如:小试、中试、商业化级别以及加样试验)
方法 4:在确信原料药中的杂质一定会低于可接受限度时(基于杂质性质以及清除知识等),建议该杂质不需要进行任何检测。
e.g.:适用于极不稳定的杂质(二氯亚砜、亚硫酰氯等);早期引入,后续被有效清除的杂质;
可以通过对工艺的控制取代分析方法的控制,
需要大量实验数据支持:中试数据积累。
3.残留溶剂的分类及限度

来源:中国药典四部附录
其中异丙基苯、甲基异丁基酮和三乙胺的限度在2020版中国药典中都进行了修订。
4、元素杂质的分类、限度及控制策略
元素杂质的分类

元素杂质的限度

来源:ICHQ3D
元素杂质的控制策略
元素杂质控制应考察检测到的元素杂杂水平相对于其PDE值的显著性。将药品中元素杂质PDE值的30%定义为控制阈值,作为元素杂质水平显著性的衡量指标。控制阈值可用于判断药品中的元素杂质是否需要额外的控制。
如果药品中某个元素杂质水平总是小于PDE值的30%,只要对数据进行了适当的评估并表明对元素杂质经行了足够的控制,则不再需要额外的控制。
如果风险评估无法表明某个元素杂质水平始终低于控制阈值,就需要建立控制方法以保证药品中元素杂质水平不超过PDE值。
5、仿制药杂质研究的一般思路
(1)杂质谱分析
基于合成工艺,分析可能产生的工艺杂质;
基于结构特征及强制降解试验,分析潜在的降解产物;
(2)原研药品的杂质分析
检索BP、JP及USP等,获取相关质量标准,得到相关已知杂质信息;
通过FDA、EMA及PMDA等,获取相关审评文件,得到相关杂质研究信息。
(3)杂质对比研究
杂质种类及杂质含量与原研进行比较;
杂质种类与原研一致或少于原研,杂质含量不超过原研;
(4)杂质限度制定
指导原则;
稳定性数据;
原研杂质情况。
结语
药品杂质的研究一直是药品研发的一项重要内容,它包括有机杂质、无机杂质及残留溶剂,这一研究贯穿于整个药品研发过程。为了能连续生产出安全、有效、质量可控的产品,在仿制药的研发过程中,以终为始,我们需要把质量源于设计贯穿始终,在各个阶段把杂质研究工作做好。
不感兴趣
看过了
取消

人点赞

人收藏

打赏
不感兴趣
看过了
取消
©2012-2023 北京华媒康讯信息技术股份有限公司 All Rights Reserved. 注册地址:北京 联系电话:010-82736610
广播电视节目制作经营许可证 —(京)字第 17437号 京海食药监械经营备20200522号
京ICP备12011723号 京ICP证150092号
 京公网安备 11010802020745号
工商备案公示信息
互联网药品信息服务资格证书((京)-非经营性-2020-0015)
京公网安备 11010802020745号
工商备案公示信息
互联网药品信息服务资格证书((京)-非经营性-2020-0015)






您已认证成功,可享专属会员优惠,买1年送3个月!
开通会员,资料、课程、直播、报告等海量内容免费看!

打赏金额
认可我就打赏我~
1元 5元 10元 20元 50元 其它
打赏作者
认可我就打赏我~
扫描二维码
立即打赏给Ta吧!
温馨提示:仅支持微信支付!
已收到您的咨询诉求 我们会尽快联系您
































 010-82736610
010-82736610
 股票代码: 872612
股票代码: 872612




