
本研究表明,数据处理工具对宏基因组和总RNA-Seq数据的影响在重复和评估水平之间存在显著差异。
编译:微科盟鼻涕,编辑:微科盟居居、江舜尧。
微科盟原创微文,欢迎转发转载,转载须注明来源《微生态》公众号。
导读
宏基因组和总RNA测序(总RNA-Seq)具有改善不同微生物群落的分类学鉴定的潜力,这可能允许将微生物纳入常规生态评估。然而,这些target-PCR-free技术需要更多的测试和优化。该研究使用672个数据处理流程处理了一个商业化微生物模拟群落的宏基因组和总RNA-Seq数据,确定了最准确的数据处理工具,并比较了它们在相同和不断增加的测序深度下的微生物鉴定准确性。数据处理工具的准确性在重复之间有很大差异。在相同甚至几乎比宏基因组学低一个数量级的测序深度下,总RNA-Seq比宏基因组学更准确。本研究结果表明,虽然数据处理工具还需要进一步探索,但总RNA-Seq可能是宏基因组学在微生物群落的target-PCR-free分类鉴定方面的有利替代方案,并可能在保持准确性的同时大幅降低测序成本。这对于常规生态评估尤其有利,因为常规生态评估需要具有成本效益但准确的方法,并且可能允许将微生物纳入生态评估。
论文ID
原名:Metagenomics versus total RNA sequencing: most accurate data-processing tools, microbial identification accuracy and perspectives for ecological assessments
译名:宏基因组与总RNA测序的比较: 最准确的数据处理工具、微生物鉴定的准确性和生态评估的前景
期刊:Nucleic Acids Research
IF:19.16
发表时间:2022.8
通讯作者:Christopher A. Hempel
通讯作者单位:加拿大圭尔夫大学
DOI号:10.1093/nar/gkac689
实验设计
将市售微生物模拟群落与超纯水混合,得到3个模拟群落重复。样品经0.2 um过滤器过滤。DNA和总RNA平行提取并进行鸟枪法测序,代表两种测序方法(宏基因组和总RNA-Seq)。测序数据使用768种常用数据处理工具组合进行处理,即数据处理工作流程。通过计算每个工作流程确定的精度指标与基于已知模拟群落组成的参考精度指标之间的欧氏距离,统计评估每个工作流程的准确性。
图1 研究设计总结。
结果
1. 测序结果
本研究获得了模拟群落样本和对照样本的2,428,038条reads(生物项目编号:PRJNA819997,SRA登录号:SRR18488964-SRR18488973)。由于我们在文库制备过程中根据体积对DNA和RNA样本进行了归一化,其中包括来自不同项目的DNA/RNA浓度较高的样本,因此在DNA和RNA文库中得到的样本的读长数取决于其他归一化样本的浓度。模拟群落样本和其他样本之间的比例在DNA和RNA样本之间似乎有很大的差异,DNA样本比RNA样本获得了更多的读长(DNA样本平均710,545 reads,RNA样本平均97,642 reads)。因此,DNA样本被随机抽样,以便在可比的读长深度下进行比较。值得注意的是,大多数RNA读长包含重复,这可能是由于相同的核糖体序列,而重复只占DNA读长的一小部分。所有阴性对照均包含少量读长,处理方法与样品相同。从样本中减去阴性对照读长以消除交叉污染,但这并不影响所呈现的结果。
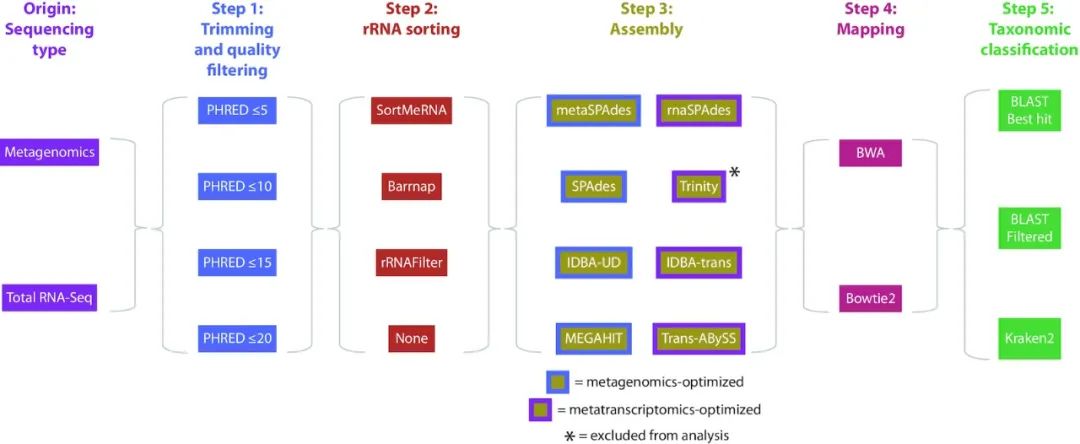
图2 应用于宏基因组和总RNA-Seq数据的768个工作流程的总结,包括用于处理数据的步骤和工具。我们无法成功地运行Trinity,并将其排除在进一步的分析之外,因此,成功运行工作流程的总数为672个。
2. 总RNA-Seq和宏基因组最准确的数据处理工具
重复和评估水平之间的结果存在很大差异,即用于评估的分类等级(种/属)和数据类型(丰度/P-A)(图3)。图3中的每一列表示三个重复中的一个所使用的评估级别,并显示了在工作流集群中具有最高平均精度的数据处理工具的相对比例、工具和精度之间的相关性的显著性以及最精确的工具。工具频率和最精确的工具分别在每一个处理步骤中进行了评估。
在最精确的集群中,工具的相对频率表明了特定工具在集群中的主导地位,由单一工具的高频率表示。使用PHRED评分和映射工具的相对频率总体上均匀分布,这表明除了一些小型集群外,最准确的工作流与使用的PHRED评分和映射工具无关。对于所有其他处理步骤,集群在大多数情况下由一到两个工具主导。然而,各重复之间的主要工具差异很大,只有少数例外,特别是Kraken2在物种丰度评估中主导了最准确的基于宏基因组的工作流集群,Barrnap在属丰度和物种-P-A评估中占据了最准确的基于宏基因组的工作流集群,BLAST在属丰度和物种-P-A评估中占据了最准确的基于总RNA-seq的工作流集群。在所有的评估水平上都无法确定装配步骤的最佳工具,并且基于宏基因组的工作流与优化的装配程序或基于总RNA-seq的工作流和/或基于总RNA-seq优化的装配程序之间也没有关系。除了少数重复外,BLAST在所有评估级别上的性能都较差。
显著性表明工具是否在所有使用的工作流中始终与更高的准确性相关,因此始终表现得更好。无显著相关性(P > 0.05)并不表示性能较差,而是表明性能也依赖于工作流中使用的其他工具。使用PHRED评分和映射工具与准确性没有显著相关性,只有少数例外。值得注意的模式是,Kraken2在基于宏基因组学的工作流程中总体上与较高的准确性显著相关,而BLAST在基于RNA-seq的工作流程中总体上与较高的准确性显著相关。
单一最准确的工具在重复和评估水平之间有所不同,但通常是占主导地位的工具。值得注意的是,对于基于P-A的评估,通常在一个步骤中执行相同的多个工具。这在使用PHRED评分和映射工具时最为显著,进一步证实了准确性与这两个步骤中使用的工具无关。Kraken2是几乎所有基于宏基因组的工作流程中精度最高的工具之一,BLAST用于基于RNA-seq的工作流程也是如此。
总体而言,最准确的工具和工作流程取决于评估水平,并且在评估水平内存在很大差异,重复之间的表现也有所不同,这表明工具的准确性和显著性可能因样本而异,即使在高度控制的模拟群落的重复之间也是如此。Kraken2和BLAST分别在宏基因组和总RNA-Seq工作流程中最适合进行分类学分类。所使用的PHRED评分和映射工具在准确性方面是可互换的。装配程序和rRNA分选工具非常依赖于评估水平和重复,然而,没有分选或Barrnap在rRNA分选方面总体上优于rRNAFilter和SortMeRNA。
图3 数据处理工具在最精确工作流集群中的相对频率(圆的大小),工具与精度之间相关性的显著性(圆的颜色),以及基于不同评估水平的最精确工具(圆中心的点)。
评估水平由测序类型(宏基因组/总RNA-Seq)、数据类型(丰度/P-A)和分类等级(属/种)组合组成。分别为每个数据处理步骤确定工具的相对频率和精度最高的工具。重复和评估水平之间的性能差异很大。
3. 比较相同测序深度下宏基因组和总RNA-Seq的准确性
所有重复在所有评估水平上都表现出差异(图4)。然而,根据到参考点的欧氏距离,在所有评估水平上,基于总RNA-seq的工作流程与参考数据的相似性显著高于基于宏基因组的工作流程(属丰度:P = 0.011,属-P–A: P = 0.014,种丰度:P = 0.019,种-P–A: P = 0.003),表明如果使用适当的数据处理工具,基于总RNA-seq的工作流程在准确性方面优于基于宏基因组的工作流程。
对于基于P-A的评估,指标和重复之间的绝对差异很小(图4)。总RNA-Seq 1的重复是最准确的,在10个预期属/种中分别检测到5个和7个(TP=5和7),仅检测到2个假阳性属和种(FP=2)。
在比较基于丰度评估的单个指标时,所有基于宏基因组的重复都引入了大量假阳性属,而这种情况只出现在基于总RNA-seq重复中的三分之一(图4)。相比之下,所有宏基因组和总RNA-seq重复都引入了大量的假阳性物种(图4)。进一步对假阳性物种的丰度和组成进行分析发现,假阳性物种大多由NA组成,即未分类的物种(平均分别占所有宏基因组和总RNA-Seq reads的23.7%和22.7%),而个别假阳性物种的贡献相对较小(平均分别占所有宏基因组和总RNA-Seq reads的0.13-1.6%和0.01-7.9%)。然而,鉴于大多数预期类群的预期丰度极低,这些结果表明假阳性物种的丰度仍然大大高于大多数预期类群。
基于丰度的指标 根据制造商提供的基因组拷贝数跨越了五个数量级(表1),所有基于宏基因组和基于总RNA-seq的重复未能检测到4-6个丰度最低的类群。值得注意的是,总RNA-Seq检测到的属比宏基因组多2个,其中一个属的丰度比未检测到的属低一个数量级。这表明丰度不是检测的唯一因素,如果一个属被检测到,其他机制也会受到影响。
基于物种丰度指标的总RNA-Seq 1只检测到最丰富的物种,Listeria monocytogenes,而没有检测到其他分类单元,这表明用于该重复的所有其他工作流程的准确性较低,因此,就与参考数据的欧氏距离而言,不检测9/10个物种比检测多个具有偏差丰度的预期分类单元更准确,对于精度较低的其他工作流程也是如此。
表1 模拟群落微生物组成。
图4 比较基于欧氏距离的多个评估水平的 每个重复的最准确的基于宏基因组和总RNA-seq的工作流程。
评估水平由数据类型组合(丰度/P-A)和分类等级(属/种)组合组成。热图中间一行的参考值代表预期指标,宏基因组或总RNA-Seq指标与参考值越接近,就越准确。所有重复在所有评估水平上均存在差异;然而,基于RNA-seq的工作流程与参考数据的相似性高于基于宏基因组的工作流程。对于基于P-A的评估,指标和重复之间的绝对差异很小(左)。基于宏基因组和总NA-seq的重复未能检测到丰度最低的5或6个类群(右)。
4. 测序深度与精度的关系
测序深度和准确度之间的关系因评估水平和测序类型而异(图5)。值得注意的是,对于基于丰度的评估,子样本到参考样本的平均欧氏距离在达到20000 reads时增加。这一观察结果是由于测序深度增加时发现了更多预期的分类群,但最初的丰度比根本没有发现预期类群时更有偏差,这最初降低了准确性。这使得多达20000 reads的子样本对于基于回归曲线的总RNA-Seq和宏基因组的比较是不切实际的,因此我们排除了它们,但各自的曲线见补充图S2。
在除P-A属外的所有评估水平上,总RNA-Seq的平均准确度比宏基因组的平均准确度提高得更快。然而,当检测总RNA-Seq重复的准确性与宏基因组重复相比是否显著更快地增加时,即回归曲线系数存在显著差异时,没有发现显著差异(属丰度: Pcoef= 0.202, 属-P–A: Pcoef = 0.573, 种丰度: Pcoef = 0.457, 种-P–A: Pcoef = 0.226)。这可能是由于重复之间的高差异(图5)。虽然不能确定具有统计学意义的差异,但观察到的回归曲线趋势表明,随着重复次数的增加,可能会出现显著的统计学差异。
尽管如此,在除物种丰度外的所有评估水平上,总RNA-Seq的平均准确度始终高于宏基因组,即使在测序深度上比宏基因组低约一个数量级。在物种丰度评估水平上,总RNA-Seq的平均准确度起初低于宏基因组,但迅速提高,直到接近相等。包含或不包含测序方法(宏基因组/总RNA-seq)作为二元自变量的线性模型之间的部分F检验证实,对于除物种丰度外的所有评估水平,模型中加入测序方法显著提高了欧氏距离预测(属丰度: Pseq < 0.001, 属-P–A: Pseq < 0.001, 种丰度: Pseq = 0.213,种-P–A: Pseq< 0.001)。这些结果证实了总RNA-Seq比宏基因组更准确。
图5 多个评估水平的测序深度与准确性的关系。
评估水平由数据类型(丰度/P-A)和分类等级(属/种)组合组成。蓝色和红色线表示所有宏基因组和总RNA-Seq重复的平均欧氏距离,每个重复下采样10次,线周围的区域表示标准差(SD)。较低的欧氏距离代表较高的精度。单个重复显示为灰色线。回归曲线显示为宏基因组和总RNA-Seq序列之间可比较的部分数据的黑色虚线。
讨论
本研究的第一个目标是测试哪一种数据处理工作流程对总RNA-Seq最准确,以及它是否与最准确的宏基因组数据处理工作流程相吻合。我们测试了736种不同的工作流程,因为许多研究强调,基于HTS的结果在很大程度上受到用于处理数据的工具的影响,我们假设不同的工具分别对宏基因组和总RNA-Seq数据最准确,因为两种测序方法会导致不同的读长组成。
本研究结果表明,对于质量筛选和修剪、rRNA排序和映射这三个步骤,宏基因组和总RNA-Seq最准确的工具是相似的,质量筛选、修剪和映射工具的选择总体上对这两种测序方法的准确性没有影响。只有在分类步骤上,最准确的工具有所不同,Kraken2总体上对宏基因组最准确,BLAST对总RNA-Seq最准确。然而,评估水平和重复之间的差异是明显的,特别是对于组装,这表明最准确的工具依赖于评估水平和重复,而不是测序类型。这些结果表明,某些处理步骤比其他步骤需要更多的关注,在我们的研究中,这将涉及到所使用的rRNA排序工具、组装器和分类器,而修剪、质量过滤和映射的选择可能需要较少的关注。值得注意的是,根据平均序列质量分数和每个序列质量分数,我们的序列质量几乎都超过了PHRED 30,因此,修剪和质量过滤可能在我们的研究中可能没有太大影响,但在产生低质量数据的研究中仍可能产生很大影响。此外,最准确的工具高度依赖于重复,这一事实表明群落在重复之间存在差异。我们研究中使用的微生物模拟群落的平均相对丰度偏差最大为30%,这意味着与理论成分的偏差是可能的。考虑到大多数模拟群落类群的丰度极低,并且在将模拟群落分为三个重复以及在过滤、提取和测序过程中可能引入了额外的偏差,因此在重复之间结果略有差异也就不足为奇了。为了比较宏基因组和总RNA-Seq最合适的数据处理工具,需要进一步模拟基于群落的测试,包括不同的分类群,更多的重复,理想情况下,重复之间的变异性更小。
据我们所知,在各种数据处理工具的测试组合范围内,我们的基准测试方法的规模是独一无二的。然而,虽然它允许我们分析单个数据处理步骤和工具对微生物鉴定准确性的影响,但它不包括对具有不同分类组成的多个数据集的测试。此外,由于我们只测试了先前研究中最受支持和最常用的工具,因此与专注于单个处理步骤的基准研究相比,每个步骤所包含的工具数量较低。这也允许测试特定工具的参数修改;例如Hleap等人采用网格搜索方法对每个工具的多个参数进行测试,选择最合适的参数,而Vollmers等人测试了两种不同k-mer长度的多个组装程序。这种微调在我们的研究范围内是不可行的,但可能会提高特定工具的性能。然而,为了确定特定处理步骤的影响,并比较宏基因组和总RNA-Seq最准确的数据处理工具,我们的基准研究在更广泛的范围内提供了重要的信息。
本研究检测到使用宏基因组数据的宏基因组优化组装器和使用总RNA-Seq数据的宏转录组优化组装器在准确性上没有差异。宏转录组优化的组装程序的开发是为了克服RNA-Seq数据之间表达水平不均匀的问题,这阻碍了低表达转录组的组装。尽管这些组装器中的许多已经被开发出来并在RNA-Seq数据上进行了测试,但只有一次使用宏基因组数据进行了相互测试。Bushmanova用RNA-Seq数据测试了他们的宏基因组优化SPAdes组装器,具有相似的性能,但倾向于错误地生成更长的contigs,并且无法检测mRNAs的亚型。基于这一点和我们自己的发现,我们建议使用已经在宏基因组特定基准测试研究中验证过的组装程序,如metaSPAdes和MEGAHIT。
基于我们的中心思想,总RNA-Seq比宏基因组恢复了更多的分类信息序列,我们的第二个目标是测试在相同的测序深度下,总RNA-Seq是否比宏基因组提供了更准确的分类鉴定。我们的结果证实了这一点,提高了应用target-PCR-free测序时提高不同微生物群落分类鉴定准确性的潜力。这反过来可以提高生态评估的有效性,因为微生物对环境变化的反应更快,因此可能比其他类群更好地代表环境条件,如原核生物、单细胞真核生物或两者所示。
本研究的第三个目标是测试随着测序深度的增加,总RNA-Seq的准确性是否比宏基因组提高得更快,这是由于恢复的总RNA-Seq reads中有更大的分类信号。虽然我们的结果不支持这一点,但总RNA-Seq的准确性与宏基因组相当,甚至在测序深度低一个数量级的情况下优于宏基因组。此外,我们的结果表明,如果我们增加重复次数,我们的假设可能会得到证实。Leese等人指出,在提出新的生态评估方法时需要考虑成本效益,尽管target-PCR-free技术具有巨大的潜力,但在常规应用中缺乏充分的验证和适当的参考数据。我们的研究表明,总RNA-Seq可能是一种受欢迎的、经济高效的、非靶向PCR的方法,值得进一步探索,因为它可以在相当低的测序深度下有效,从而大幅降低成本。传统的宏转录组学在生态评估方面也得到了越来越多的关注。有人认为微生物功能可能比分类单元的身份更能反映环境的变化,因为功能转移可能发生在分类学转移之前。对宏转录组学日益增长的兴趣也可能增加总RNA-seq的适用性,因为两种方法可以使用相同的RNA提取物进行互补。这将有一个额外的好处,即能够研究分类群-功能的关系。然而,由于RNA的不稳定性和RNA样本采集成本较高,将RNA纳入常规生态评估的适用性也受到了关注。但最近对环境RNA (eRNA)的研究表明,它在环境中的稳定性要比之前认为的高得多,这使得它确实适合于常规生态评估,并为一个全新的环境研究领域(环境转录组学)提供了可能性。
结论
本研究表明,数据处理工具对宏基因组和总RNA-Seq数据的影响在重复和评估水平之间存在显著差异。此外,宏基因组优化的组装程序不能统一地改善宏基因组数据,宏转录组优化的组装程序也不能统一地改善总RNA-Seq数据。需要对特定数据处理步骤进行更高分辨率的进一步研究,以便为给定的背景调整最合适的选择。与宏基因组学相比,总NA-Seq在测序深度相同的情况下,甚至在测序深度低一个数量级的情况下,为微生物模拟群落提供了更准确的分类鉴定。这些结果表明,在对微生物群落进行target-PCR-free分类鉴定时,总RNA-Seq是宏基因组学的一个很好的替代选择,在保持准确性的同时可能大幅降低测序成本。这可能有利于常规生态评估,因为常规生态评估需要具有成本效益的方法,并允许将微生物纳入生态评估。在当前eRNA和环境转录组学研究的背景下,总RNA-Seq可能是宏转录组学的一种补充方法,允许建立分类群-功能关系。需要对环境样本进行进一步研究,以确认总RNA-Seq在应用环境中优于宏基因组的优势。
不感兴趣
看过了
取消

人点赞

人收藏

打赏
不感兴趣
看过了
取消
©2012-2023 北京华媒康讯信息技术股份有限公司 All Rights Reserved. 注册地址:北京 联系电话:010-82736610
广播电视节目制作经营许可证 —(京)字第 17437号 京海食药监械经营备20200522号
京ICP备12011723号 京ICP证150092号
 京公网安备 11010802020745号
工商备案公示信息
互联网药品信息服务资格证书((京)-非经营性-2020-0015)
京公网安备 11010802020745号
工商备案公示信息
互联网药品信息服务资格证书((京)-非经营性-2020-0015)






您已认证成功,可享专属会员优惠,买1年送3个月!
开通会员,资料、课程、直播、报告等海量内容免费看!

打赏金额
认可我就打赏我~
1元 5元 10元 20元 50元 其它
打赏作者
认可我就打赏我~
扫描二维码
立即打赏给Ta吧!
温馨提示:仅支持微信支付!
已收到您的咨询诉求 我们会尽快联系您





































 010-82736610
010-82736610
 股票代码: 872612
股票代码: 872612




