

科研丨GENOME BIOL: 宏基因组和宏转录组数据综合分析揭示阴道细菌生态



编译:微科盟北岸,编辑:微科盟居居、江舜尧。
微科盟原创微文,欢迎转发转载,转载须注明来源《微生态》公众号。
导读
以乳杆菌物种为主的阴道菌群与各种不良健康后果的风险降低有关。然而,许多健康女性的微生物群并不以乳杆菌为主。为了确定推动阴道菌群组成的因素,对健康女性阴道菌群的基因组成和转录活性进行了表征。结果表明,一个物种的丰度并不总是其转录活性的指示物,并且可以从转录组学数据预测群落组成的未来变化。功能比较突出了这些群落代谢活动的差异,尤其是在降解宿主产生的粘蛋白(而非糖原)方面。非乳杆菌主导的菌群对粘蛋白的降解可能与不良健康结果有关。最后,本研究发现L. crispatus、L. iners和Gardnerella vaginalis的转录活性随其所在群落的分类组成而变化。值得注意的是,L. iners和G. vaginalis与乳杆菌共存时均表现出较低的胆固醇依赖性溶细胞素表达,而当与其他兼性和专性厌氧菌共存时表现出较高的表达。这些物种的致病潜力可能取决于它们所存在的菌群,因此可以通过干预策略进行调节。本研究为阴道菌群的功能生态提供了见解,展示了宏转录组数据的诊断潜力,并揭示了这些生态系统的调节策略。
论文ID
原名:Insight into the ecology of vaginal bacteria through integrative analyses of metagenomic and metatranscriptomic data
译名:宏基因组和宏转录组数据综合分析揭示阴道细菌生态
期刊:Genome Biology
IF:17.906
发表时间:2022.3
通讯作者:Jacques Ravel
通讯作者单位:美国马里兰大学医学院
DOI号:10.1186/s13059-022-02635-9
实验设计

结果
1 物种的相对丰度并不一定表明其对宏转录组的贡献
研究人员对39名北美育龄妇女(年龄范围:19-45岁)在5个时间点(间隔2周)的阴道微生物群进行了表征。研究中的受试者是黑人或非裔美国人(n =24)、白人或高加索人(n = 10)、西班牙裔或拉丁裔(n = 4)和亚裔(n = 1)。对这些样本进行宏基因组(n = 194)和宏转录组(n = 180)测序,并将测序结果映射到VIRGO,这是一个非冗余的、全面的阴道微生物组基因目录,以确定宏基因组和宏转录组的分类组成(图1)。宏基因组组成代表菌群的相对丰度,而宏转录组组成代表菌群的相对活性(附表1)。 研究人员首先确定一个物种的相对丰度是否与其相对转录活性一致。一个物种的相对活性除以其相对丰度的比率被称为其“相对表达”,代表了与宏基因组相比,该物种在宏转录组中的表达是过度或不足。研究人员计算了25种高丰度的菌种在研究范围内的相对表达值,其中包括四种常见的阴道乳杆菌物种,G. vaginalis、Atopobium vaginae、Ca.Lachnocurva vaginae和Prevotella spp.,以及其他几种兼性或专性厌氧菌(图2A)。对于每个物种和样本,观察到的相对表达值范围很广,从过度表达到表达不足。G. vaginalis、A. vaginae、Finegoldia magna、Mobiluncus mulieris和咽峡炎链球菌(Streptococcus anginosus)更可能表达不足(中位相对表达< 0,图2A),而其他20种更可能表达过度(中位相对表达 > 0),尤其是Sneathia amnii(中位相对表达= 3.3,图2A)和S. sanguinegens(中位相对表达= 2.2,图2A)。
研究人员分析一个物种的相对表达是否与其相对丰度相关。因为这两个数据集都是组成的,所以对相对表达的评估以最高和最低相对丰度为界。如果估计一个物种的相对丰度为90%,其相对表达的最大值约为1.1。相反,如果一个物种只占菌群群落的一小部分,其相对表达的最小值受转录组测序深度的限制。研究人员发现一个物种的丰度与其相对表达之间存在密切关系(图2B-G;线性混合模型;F1,4261 = 7167,p < 0.001)。在极少的情况下,阴道细菌的转录活性比它们的相对丰度所显示的要高。而相对丰度较高的物种往往对宏转录组贡献不足。从过度表达到表达不足因物种而异(图2B-G;补充文件2:附图1;线性混合模型;F24,4261 = 198,p < 0.001)。例如,L. crispatus、L. iners和G. vaginalis(图2B - D)等物种往往在较高的相对丰度下被发现,一般为过度表达。相反,A. vaginae和Ca. L. vaginae菌种(图2E,F)在中等或较低的相对丰度下发现,较低的相对丰度下活性表达不足(x-截距 < 10−2相对丰度)。S. amnii(图2G)和S. sanguinegens(补充文件2:附图1)相对丰度较低,但发现它们有较高的转录活性。

图1 通过鸟枪法宏基因组学评估39名女性在五个时间点的阴道菌群组成(n=194,图A)。
每个物种对相应样本相对表达活性的贡献(n=180,图B)。这两个估计值都是使用VIRGO得出的,并对基因长度进行了校正。样本在两个数据集中按相同的顺序排序。缺口表示没有相应的宏转录组数据可用的14个样本。基于VALENCIA的分类组成,将群落划分为CST:CST I–L. crispatus为主,CST II–L. gasseri为主,CST III–L. iners为主和CST V–L. jensenii为主和CST IV。

图2 物种的表达活性并不总是与其相对丰度相匹配。
阴道菌群中25个最丰富物种的相对基因表达是通过将物种相对表达活性除以其在宏基因组中的相对丰度(A)来计算的。物种在群落中的相对表达与其相对丰度之间的关系(B-G)。
分类单元的相对表达活性可以预测其未来丰度的变化
一个物种或菌株的相对转录活性可能对其未来在微生物群落中的丰度有一定的影响。人们可能会认为,相对表达量低的物种可能会经历相对丰度的下降,而相对表达量高的物种可能会增加。此研究的纵向数据集能够验证这一假设。研究人员评估了间隔两周的两个连续时间点之间log10转化物种相对丰度的变化。正如假设的那样,此研究发现一个物种的相对丰度变化与其早期表达水平之间存在密切关系(图3A;线性混合模型;F1,1847 = 649,p < 0.001)。相对表达低的物种相对丰度降低,而相对表达高的物种相对丰度增加。研究人员还发现在这种关系中支持物种特异性截获(图3B;线性混合模型;F24,1847 = 11.1,p < 0.001),这代表了物种在2周时间间隔内维持其相对丰度所需的相对表达。大多数菌群需要增加表达,以维持其相对丰度。值得注意的是,S. anginosus和F. magna被发现能够在表达不足的情况下保持其相对丰度,而S. amnii和S. sanguinegens需要高度表达才能保持其相对丰度。
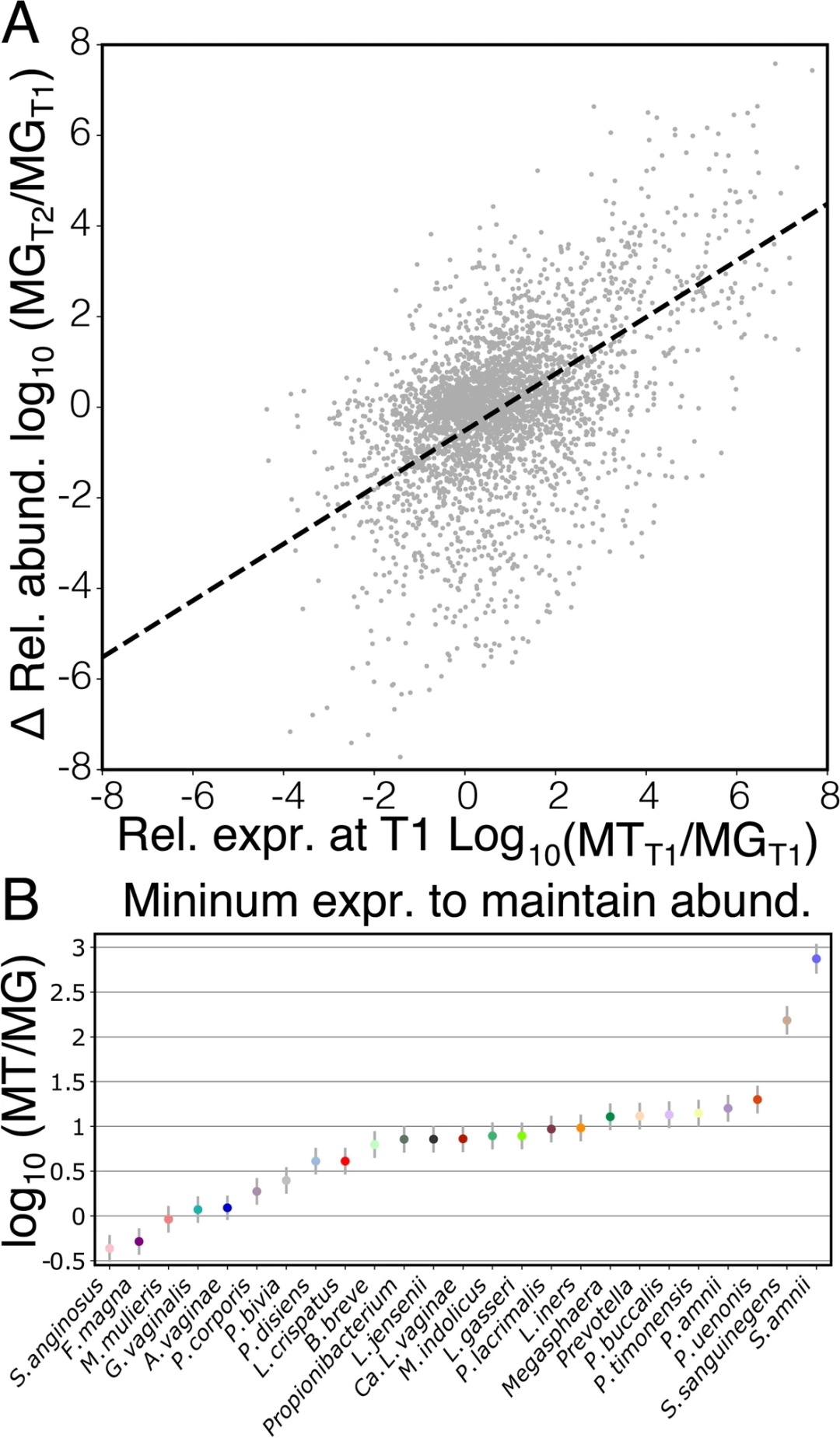
图3 菌群的相对基因表达可以预测其相对丰度的变化。
一个物种从一个时间点(T1)到下一个时间点(T2)的相对丰度的Log10倍变化,作为其在T1时相对表达量的函数(A)。该模型中的物种特异性截获代表物种在T1至T2之间维持其相对丰度所需的最小相对表达(B)。
3 不同CSTs菌群转录活性的功能差异
根据定义,CSTs在组成方面有很大差异,因此,预期它们在功能方面也会有很大差异。为了表征这些差异,研究人员在代表分子功能的KEGG orthologs(KOs)水平上进行了差异表达分析。将分析重点放在CST I、CST III和CST IV的比较上,因为该数据集中几乎没有CST II和CST V的例子。这些CSTs之间的比较揭示了大量差异表达的KO(图4A-C)。其中包括预期的差异,如CST I中D-乳酸脱氢酶(K03778)的表达量比CST III和IV高4.6倍和4.8倍,其催化丙酮酸产生D-乳酸。在CST I和IV之间,KO差异表达的数量和幅度最大(图4B)。在宏基因组数据中,大多数被鉴定的差异表达KEGG功能差异丰富,表明这些群落的功能能力存在潜在差异。然而,也有一些KO同样丰富,但表达不同(图4A-C中用黄色钻石表示)。
例如,ptsAB磷酸转运系统(K02036和K02038)在CST I、III和IV中的丰度相等,在CST I中的表达比在CST III和IV高约4倍。另一个例子是α-L-鼠李糖苷酶(K05989),其在CST I和IV中的丰度相等,但在CST IV中的表达高3倍。 接下来,我们表征了对每个差异表达的KO有贡献的分类群,以确定CSTs活性中功能变异的驱动因素(图4D-F)。对于每个KO,计算了每个分类单元对其在每个CST中表达的平均贡献。正如预期的那样,研究人员发现,与CST III或CST IV相比,CST I中富集的KEGG功能主要来源于L. crispatus(图4D,E)。因为,L. crispatus是CST I的优势物种。与CST I相比,CST III中丰富的功能来源并不主要是CST III的优势菌群(L. iners)。
相反,这些功能的表达主要来源于G. vaginalis、短双歧杆菌(Bifidobacterium breve)和几种Prevotella spp.(2-4倍差异,图4D)。只有KO在这个比较中显示出较高的倍数变化,主要归因于L. iner。与CST I或CST III相比,发现在CST IV中富集的KO的表达可归因于多个物种。G. vaginalis和A. vaginae是CST IV群落的两个特征类群,但它们并不负责这些差异表达的KO中的大多数;而Ca. L. vaginae、Sneathiaspp.和Prevotellaspp.则表达这些功能。 差异表达的KO也被分组到KEGG模块。这种更高层次的功能分类描述了KOs,它们协同作用以执行功能。基于KEGG模块与CST I、III和IV的关联模式,使用层次聚类对其进行分组。这将KEGG模块区分为7组(图5)。
其中一组包括在CST I中表达高于CST III或IV的KEGG模块。这一组包括β-葡萄糖苷、甘露醇、膦酸酯、磷酸盐和腐胺转运系统,以及半胱氨酸、赖氨酸、胡萝卜素和辅酶F420的生物合成。另一组KEGG模块与CSTs I和III中的高表达相关,包括锌/锰、谷氨酰胺、葡萄糖醇转运体,以及熊果苷样PTS系统。没有一组KEGG模块在CST III中表现出单独高表达,这可能反映了L. iners因其基因组大小减小而导致的功能有限。相反,有两组模块在CST III和IV中表现出较高的表达,这些模块与CST III的关联主要由除L. iners以外的物种驱动。这两组包括谷氨酸转运、蛋氨酸降解、铁和锌转运以及组氨酸生物合成。最后,一些模块仅与CST IV相关。该组包括负责支链氨基酸转运、麦芽糖/葡萄糖运输、细胞溶血素运输、趋化性、半乳糖胺运输、LPS运输和硫降解的KEGG模块。

图4 CST转录活性和相关分类驱动因素的功能差异。
火山图显示了不同群落状态类型(A、C和E)之间KEGG orthologs表达的log2倍变化差异。蓝色点代表差异表达和差异丰度的KEGG orthologs。黄色点表示差异表达但丰度无差异的KEGG orthologs。在B、D和F中,显示了负责这些KO表达的分类群。对于每个倍数变化类别,将显示每个分类单元的相对贡献。条形图上方的数字表示该类别中差异表达的KEGG orthologs的数量。*DE,差异表达。

图5 与CST相关的KEGG模块表达差异相关。差异表达的KEGG同源基因被映射到KEGG模块(代表代谢功能)。
热图显示了KEGG模块的平均对数2倍变化值,在三组比较(CST I与CST III;CST I与CST IV,或CST III与CST IV)中的一次比较中显示出至少四倍的差异。三组比较中的每一次都绘制了两列,每一列代表KEGG模块,其直系图在指定的CST中表现出更高的表达。一些KEGG模块包括KEGG同源基因,它们在每个比较的CST中都有较高的表达。使用Euclidean距离和Ward链接对倍数变化值进行分层聚类,将模块分为七类。
4 阴道菌群糖原和粘蛋白利用基因的表达
宿主产生的糖原和粘蛋白都被认为是阴道细菌碳和能量的重要来源。最近的研究已经确定了微生物群代谢这些产物的相关基因,包括Lactobacillus spp.的糖原脱支酶和G. vaginalis的唾液酸酶。本研究检测了这些酶和其他参与糖原和粘蛋白降解的酶,以确定它们在分配给不同CSTs的菌群中的表达是否不同。
使用碳水化合物活性酶(CAZy)注释确定了负责这些功能的VIRGO基因。糖基水解酶(GH)家族13和糖基转移酶(GT)家族35中的酶可能分别具有糖原脱支和糖原磷酸化酶活性。脱支酶作用于α-糖苷键,可以从糖原中释放海藻糖、葡萄糖、麦芽糖和麦芽三糖。这些酶在CST中的表达相似,约为每百万1250个转录本(TPM),尽管在CST III菌群中的表达略高(线性混合模型;图6A)。糖原脱支酶的表达归因于阴道常见的许多细菌,包括Lactobacillus spp.、G. vaginalis、Ca. L. vaginae、Prevotella spp.和Sneathia spp.,并且与CSTs的物种组成一致。
例如,L. crispatus负责CST I中大部分糖原脱支酶的表达(图6B)。糖原磷酸化酶的表达因CSTs而异,其作用是从糖原中切割单个葡萄糖单体,其表达在CST IV中最高,在CST I中最低(线性混合模型,图6C)。这可能是由于在CST I和III中占主导地位的Lactobacillus spp.基因组中缺少这些基因。负责糖原磷酸化酶表达的分类群在CSTs中相似(图6D)。
宫颈细胞产生的粘蛋白被各种碳水化合物残基广泛修饰,释放后可被微生物群代谢。从这些粘蛋白连接的低聚糖中去除末端残基,通常是唾液酸或岩藻糖,通常是它们降解的第一步。负责去除唾液酸和岩藻糖的酶分别是唾液酸酶(GH家族33)和岩藻糖苷酶(GH家族29)。两种酶家族的表达在CST IV菌群中最高(图6E,F),在CST I菌群中最低,这表明当Lactobacillus spp.在阴道微生物群中占优势时,粘蛋白代谢可能较低。在所有CSTs中负责唾液酸酶表达的分类群包括G. vaginalis、一定数量的Prevotella spp.,以及短双歧杆菌的少量贡献(图6F)。相比之下,岩藻糖苷酶的表达主要来源于G. vaginalis、P. amnii、P. bivia、P. timonensis和Propionibacterium(图6H)。
半乳糖是粘蛋白的另一种常见成分,可通过α-半乳糖苷酶(GH家族36)或β-半乳糖苷酶(GH家族2)的作用释放。α-半乳糖苷酶在CST I和IV群落中的表达高于CST III群落,而β-半乳糖苷酶在CST IV群落中的表达仅高于CST III群落(图6I,K)。L. iners在这两个组中都不表达,这解释了为什么这两个酶家族在CST III菌群中表达最低。表达这些酶的非乳杆菌包括G. vaginalis、S. anginosus(仅α-半乳糖苷酶)、S. sanguinegens(仅β-半乳糖苷酶)和各种Prevotella spp.。
值得注意的是,S. sanguinegens表达了大量的β-半乳糖苷酶。总之,这与粘蛋白在CST IV菌群中降解最高一致,而在CST I和III菌群中降解有限,因为这些CST缺乏唾液酸酶和岩藻糖苷酶的表达。 本研究检测的最后一类粘蛋白利用基因是硫酸酶,其作用是从粘蛋白上切割硫酸基团,从而进一步去除碳水化合物残基。该酶占群落转录活性的0.2%,在CST I中表达最高,在CST III中表达最低(图6M)。广泛的分类群负责其表达,包括L. crispatus、L. iners和G. vaginalis(图6N)。

图6 粘蛋白的表达因CST而异,但糖原降解相关基因的表达不受CST的影响。
阴道内两种丰富的宿主源性资源(糖原和粘蛋白)与代谢相关关键功能的表达差异。左边图(A、C、E、G、I、K和M)显示了特定酶的组合转录。对于每一类酶,使用线性混合模型测试其基因表达在CST之间的显著差异,受试者作为随机因素。右边图(B、D、F、H、J、L和N)显示了每个CST成员对这些基因转录的平均相对贡献。每个受试者的每种酶类表达水平的纵向表示可在补充文件2:补充图2中找到。*p < 0.05,**p < 0.01,***p< 0.001。
5 整合宏基因组和宏转录组数据确定潜在的物种相互作用
在上述分析中,重点描述了微生物群总转录输出。考虑到序列数据集的深度和阴道微生物群表现出较低群落均匀性的趋势,研究人员还能够检测单个物种的转录活性。L. crispatus(n=48)、L. iners(n=74)和G. vaginalis(n=58)的转录组在至少30个宏转录组中有足够的覆盖率(目标物种>50万reads)。研究人员通过对宏基因组数据的综合分析,确定这些物种的基因表达是否受其群落组成的影响,并通过宏转录组数据确定重点物种的基因表达。稀疏典型相关分析用于确定这两个数据集之间的协方差轴,代表来自重点物种的基因组合,其表达与群落中物种组合的相对丰度相关。
为了克服基因序列和基因含量在同一物种不同菌株之间的差异,评估了VIRGO中阴道同源蛋白组(VOGs)的表达水平。 对于L. crispatus,研究人员确定了41个VOGs,其基因表达水平与A. vaginae和G. vaginalis的相对丰度相关(ρ = 0.80),表明这两个物种的丰度可能影响L. crispatus的转录活性。对41个VOGs和两个分类群的主成分分析(PCA)表明,A. vaginae和G. vaginalis的组合相对丰度沿第一轴变化(图7A)。其中19个呈正相关关系,较高的基因表达与A. vaginae和G. vaginalis的相对丰度增加有关,22个呈负相关关系,较高的基因表达与A. vaginae和G. vaginalis的相对丰度减少有关。
正相关关系的VOGs包括:两个碳水化合物调节基因glpR和cggR、铁和镁转运基因、ulaG维生素C利用基因、替代sigma因子sigL以及膜相关的asp23碱性休克反应基因。负相关关系的VOGs包括:羧酸酯酶nlhH、乳糖操纵子阻遏物purR、氨基酸和磷酸转运体、dnaK伴侣蛋白和谷氨酰胺合成酶基因glnA。 研究人员对L. iners转录活性的分析揭示了一组111个VOGs,它们与十个物种的相对丰度相关(ρ = 0.84),2个负相关(L. jensenii和F. magna)和8个正相关的BV(细菌性阴道病)相关类群(P. amnii、Megasphaera、A. vaginae、P. timonensis、Prevotella spp.、G. vaginalis、P. buccalis和Mageebacillus indolicus)。这一结果表明,这些VOGs的基因表达与正相关物种和负相关物种的比例相关,并且可以在相关VOGs和物种组合数据集的主成分分析中看到(图7B)。
在与L. jensenii和F. magna相对丰度较高相关的58个L. iners的VOGs中,包括氨甲酰磷酸合成酶基因(小链和大链成分)、半乳糖导入和变位酶基因、细胞形状控制rodZ基因、细胞壁生物合成基因murE/murF、依赖NH(3)的NAD(+)合成酶基因以及DNA损伤修复系统的几个组成部分(mutL、mutS2、recJ和recR)。另一方面,在八种BV-相关微生物存在的情况下,53个L. iners的VOGs表现出较高的表达,包括甘露糖/果糖/N-乙酰半乳糖胺转运基因、胆固醇依赖性溶细胞素、编码假定粘蛋白结合蛋白的基因、葡萄糖胺激酶gspK和半乳糖-6-磷酸异构酶lacAB基因。
最后,研究人员用同样的方法检测了G. vaginalis的转录活性。研究人员分析表明,一组185个G. vaginalis的VOGs可能与9种细菌物种的相对丰度相关(ρ = 0.80)。在这九个分类群中,5个被确定为正相关(L. crispatus、L. jensenii、L. gasseri、S. epidermidis和Corynebacterium pseudogenitalium),4个被确定为负相关(P. amnii、P. timonensis、M. indolicus和Megasphaera)。包含相关基因和分类群的组合数据集的PCA分析表明,样本在第一轴上正、负分类群的组合相对丰度比率有所不同(图7C)。与研究人员在L. iners中观察到的类似,相对丰度较高的G. vaginollis胆固醇依赖性溶细胞素是与BV-相关微生物相关的VOGs。与这四种BV-相关微生物相对丰度较高相关的128种VOGs包括:维生素K环氧化物还原酶基因、几种金属转运蛋白基因、内肽酶基因、丝氨酸蛋白酶基因、麦芽糖转运蛋白基因和两个可能编码糖原脱支酶和α-淀粉酶结构域的基因。
另一方面,研究人员发现57个VOGs的表达与更高相对丰度的L. crispatus、L. jensenii、L. gasseri、S. epidermidis和Corynebacterium pseudogenitalium相关。其中包括hsp70,一种groEL样伴侣蛋白,两个与葡萄球菌基因ebh相似的基因,两个麦芽糖转运基因,两个假定的糖原脱支酶,以及烷基过氧化氢还原酶基因。

图7 菌群组成调节物种基因表达。
通过稀疏典型相关分析(sCCA)对VOGs与菌群的主成分双标图进行了分析。对三种最常见物种的转录活性进行了分析:L. crispatus(A)、L. iners(B)和G. vaginalis(C)。每个图中的大圆点代表样本。在A中,这些点根据两个负贡献类群(A. vaginae和G.vaginalis)的组合相对丰度log10进行着色。在B和C中,这些点根据对相关性做出正或负贡献的分类群相对丰度的比率进行着色。图中包括基因(正,蓝色;负,洋红)和分类群(正,绿色;负,金色)的因子负荷。
讨论
本研究对阴道宏转录组和宏基因组的综合分析表明,一个物种的相对丰度并不一定反映其转录活性。在相对丰度较低时观察到的分类群在宏转录组中的代表性通常较高,而在相对丰度较高时观察到的分类群在宏转录组中的代表性通常较低。没有理由怀疑这些差异是由生物信息学处理造成的,因为宏基因组学和宏转录组学数据集是以相同的方式处理的。可能是提取DNA和RNA的技术的差异导致了观察到的差异。然而,该研究认为在这种情况下不太可能,因为发现宏基因组和宏转录组的组成之间的差异可以预测群落动态。相反,认为这种不一致性是由群落中物种之间的差异转录活动造成的。这种解释对如何看待这些菌群具有重要意义。如果估计群落中稀有的物种具有高度的转录活性,它们可能会对宿主组织产生不成比例的影响。
例如,S. amnii和S. sanguinigens在宏转录组中几乎总是过度表达,而且通常高达几个数量级。这两个物种被许多人认为是阴道病原体,并已被证明与阴道炎症有关。一些研究进一步将这些物种的丰富与自然早产风险的增加联系起来。通过检测群落的转录活性,这些物种与不良健康结果之间的关联可能会得到加强,因为当Sneathia spp.的相对丰度较低时,基于DNA的成分决定因素可能会低估其重要性。
本研究发现一个物种的转录活性可以用来预测分类群相对丰度的变化。在下一个时间点,转录组中代表性不足的物种的相对丰度通常会降低,而代表性过高的物种的相对丰度往往会增加。考虑到时间点相隔大约两周,而且这些菌群可以经历快速变化,这个结果有点令人惊讶。RNA的化学稳定性不如DNA,因此可以更准确地反映采样时的群落。当来自死亡细胞的DNA被降解或被冲出阴道时,基于DNA的成分估计也可能会经历一段滞后时间。因此结果表明,群落组成的变化比我们基于DNA的估计更早发生。这对理解推动阴道微生物群变化的因素有着深远的意义。例如,有症状的细菌性阴道病可能是在发病前几天开始的微生物过程的表现。试图识别BV的根本原因或开发新的BV诊断将受益于在BV出现之前仔细观察微生物群的转录活动。 接下来,研究人员研究了分配到不同CSTs的菌群转录活性的功能差异。根据定义,分配给不同CSTs的群落具有不同的分类组成,因此预计它们的功能活性会有所不同。然而,CSTs之间表达不同的功能代表了代谢途径,乳杆菌可以利用这些代谢途径来控制阴道微生物群的组成。
例如,CST I和III群体表达的转运体能够转运谷氨酰胺、谷氨酸和天冬氨酸,而CST IV群体表达的转运体只能转运谷氨酸。谷氨酰胺是一种肽聚糖前体,在寡肽交联中作为胺供体将果糖-6-磷酸转化为氨基葡萄糖-6-磷酸,并将天冬氨酸转化为天冬酰胺。基于这一知识,我们认为补充谷氨酰胺可能有利于阴道环境中的乳杆菌,而不是CST IV中常见的BV相关生物。这一点得到了结果的支持,即L. crispatus在与G. vaginalis 和A. vaginae共存时表现出谷氨酰胺合成酶表达增加。本研究还发现,预计可转运锌和锰的金属转运体在CST I和CST IV中的表达高于CST IV。
之前的研究表明,乳杆菌利用锰来防御氧毒性,补锌已被证明可以改善益生菌植物乳杆菌菌株的生长。 同时发现,以L. crispatus为主的群落也表现出较高的磷酸盐和膦酸酯转运体表达,这与之前的基因组比较一致,该比较表明,与L. iners相比,L. crispatus具有更多的磷酸盐/膦酸酯转运体,并且之前观察到,当阴道pH值小于4时,阴道微生物群中的磷酸盐转运基因丰度增加。磷酸盐可以缓冲pH值和多聚磷酸盐的细胞内积累,已被其他乳杆菌用作耐酸机制。
尽管尚未证明L. crispatus能产生多聚磷酸盐,但简单地隔离磷酸盐可能会使L. crispatus提高细胞内pH值并降低细胞外pH值。同样,我们发现L. crispatus优势菌群表现出较高的腐胺/精胺转运体表达,这与代谢组学研究的结果一致,L. crispatus主导的腐胺水平较低,亚精胺水平较高。控制这些pH缓冲化合物的细胞内和细胞外浓度可能是L. crispatus利用的一种竞争策略,它可以抑制竞争对手的生长并促进自身的代谢。 作为功能分析的一部分,研究人员重点研究了预计参与糖原和粘蛋白降解的酶的表达,这是阴道环境中产生的碳和能量的两种潜在来源。糖原主要由阴道上皮细胞产生,而粘蛋白主要由宫颈组织产生,然后作为粘液的一种成分分泌到阴道。阴道微生物群降解糖原的能力已经争论了一段时间,但最近的研究表明,许多阴道细菌可能确实具有降解宿主产生的糖原的能力。
研究人员证明,阴道微生物群中最常见的成员在体内表达预测的糖原降解酶,尽管需要体外研究来验证其活性。这些酶的表达在分配给不同CSTs的群体中似乎没有很大差异,这表明糖原降解在这些女性中可能以基本相同的速率发生,并且糖原是阴道微环境中的一种普遍资源。因此,一些人建议单独补充糖原治疗BV,这似乎不太可能有利于乳杆菌,实际上可能有利于BV相关细菌的生长。这一挑战可以通过用pH值降低剂补充糖原来克服,以使耐酸性更强的乳杆菌在这种常见物质的代谢中具有优势。
阴道粘液具有非特异性抗菌特性,乳铁蛋白、溶菌酶和免疫球蛋白等分子通常与之结合。宫颈阴道粘液在生育和预防包括HIV在内的性传播感染方面起着关键作用。粘蛋白似乎被阴道微生物群不同程度地降解。阴道Lactobacillus spp.不表达唾液酸酶或岩藻糖苷酶,这意味着它们可能无法从粘蛋白糖基化链中去除末端唾液酸或岩藻糖残基。在CST IV中普遍存在的分类群,包括G. vaginalis和Prevotella spp.,确实表达这些酶。Gardnerella经常出现在阴道粘蛋白降解的讨论中,但结果表明,Prevotella spp.尽管在CST IV群落中相对丰度通常较低,但也可能是Prevotella spp.活性过大的原因。研究人员得出结论,粘蛋白降解的程度和方式可能因群落组成而异。对于阴道粘蛋白糖基化链在时间和个体之间的变化,已经有一些直接的评估。
然而,这些结果很难解释,因为我们对任何微生物干扰之前的粘蛋白组成的变化知之甚少。因此,考虑到粘蛋白在生育和性传播感染易感性中的关键作用,微生物群驱动的粘蛋白组成差异可能对生殖健康产生重要影响。 在该研究最终分析中,检测了L. crispatus、L. iners和G. vaginalis的转录活性。研究人员使用宏基因组和宏转录组数据的综合分析来判定是否存在属于这些物种的基因集,其表达与群落中分类群的相对丰度相关。这些分析的结果为这些物种在阴道环境中的生态提供了功能性见解,并暗示了微生物群成员之间可能存在的相互作用。
对于L. crispatus,发现一组基因的表达与阴道A. vaginae和G. vaginalis的相对丰度相关,这两个物种在阴道环境中与L. crispatus竞争。在一组直接相关的L. crispatus基因集中包括asp23,它编码一种碱性休克蛋白。该基因的高表达可能有助于L. crispatus在较高pH条件下生长。其他相关基因(例如,碳水化合物代谢调节因子、氨基酸和磷酸盐转运体、谷氨酰胺合成酶)可能表明,当L. crispatus与这些细菌共存时,其代谢活动发生了变化。当与A. vaginae和G.vaginalis共同存在时,其转录活性变化的额外体外研究可以为调节以L. crispatus为主微生物群的状态提供策略。
研究人员对L. iners和G. vaginalis转录组的分析结果显示了许多共同点,这可能反映了它们在生态学上的相似性。当L. iners和G. vaginalis与CST IV相关的生物共存时,L. iners和G. vaginalis在基因表达上均表现出差异。L. iners通常与CST IV相关(包括G. vaginalis)的细菌共存。对于L. iners,一组相关的乳杆菌仅包括L. jensenii,该物种通常与L. iners共存。对于G. vaginalis,相关的乳杆菌包括L. gasseri、L. jensenii和L. crispatus。这些物种的相对丰度较高与G. vaginalis表达的烷基过氧化氢还原酶增加有关。L. crispatus、L. gasseri和L. jensenii在体外产生H2O2,宏转录组学数据表明L. crispatus在体内表达能够产生H2O2的酶。
过氧化氢对许多阴道常见的厌氧菌有毒,这导致许多人认为过氧化氢在乳杆菌提供的保护中起主要作用,尽管这一点受到了质疑。反对其相关性的论据包括,目前尚不清楚阴道内生理氧浓度下是否可能产生H2O2,以及H2O2不太可能在阴道内扩散很远。此结果与Gardnerella在与这些乳杆菌共存时对过氧化物具有更强的防御能力相一致,这可能会将乳杆菌产生的H2O2的影响降至最低,并提供了反对H2O2作为抗菌药物的另一个论据。Gardnerella也有可能对环境氧气浓度升高做出反应,这也可能有利于乳杆菌。这是一个有趣的发现,调节阴道微环境的氧化还原电位可以有利于乳杆菌。 本研究使用稀疏典型相关分析对L. iners和G. vaginalis转录组进行分析的第二个相似之处涉及它们的胆固醇依赖性溶细胞素。对这两个物种的比较基因组研究表明,这两个物种都可能通过水平基因转移获得溶细胞素,因为它们不存在于其他乳杆菌物种或双歧杆菌科的其他成员中。
事实上,系统发育分析表明,惰性蛋白酶与阴道溶解素最为相似,尽管最近的表型比较显示了它们之间的一些差异。在两项分析中,溶细胞素的高表达与CST IV常见的几种BV-相关厌氧菌的相对丰度较高相关,尽管两个物种的相关性中仅包括M. indolicus。这一结果表明,当存在这些微生物时,L. iners和G. vaginalis表现出较高的溶细胞素表达,而当与其他乳杆菌物种共存时,其表达较低(L. jensenii与L. iners;L. jensenii、L. gasseri、L. crispatus与G. vaginalis)。胆固醇依赖性溶细胞素通过在宿主细胞膜上形成的孔发挥作用,并已被证明有助于许多细菌的致病性,尽管鉴于该细菌尚未明确显示与不良健康状况相关,溶细胞素的致病性尚不清楚。然而,L. iners和G. vaginalis的致病潜力似乎受到其所在菌群的整体组成的调节。以L. iners为主的群落对宿主组织的破坏性可能不如包含L. iners的群落与其他BV相关物种共同存在的群落。
结论
许多微生物组研究的一个共同局限性是,它们依赖于基于DNA的群落组成评估。这些估计,无论是来自16S rRNA基因扩增子还是宏基因组学,都代表着基因组和功能潜力。并非基因组上的所有基因都在同一时间或以相同的速率转录。宏转录组测序描述了体内微生物群落的转录活性,并能更好地了解这些群落的功能。然而,宏转录组学并不能提供微生物群落生物化学活性的完整图像。mRNA稳定性和翻译效率的差异会导致每个转录本产生的蛋白质数量发生变化,蛋白质的稳定性也会不同。如果转录本编码一种酶,其活性也将受到底物、产物和辅因子浓度以及环境条件的控制。然而,宏转录组学确实在很大程度上弥合了功能潜能和已实现的生化活性之间的很大一部分差距。阴道微生物组与健康结果之间的流行病学关联应该通过检测阴道宏转录组来加强。本研究对阴道宏转录组预测能力的论证也可用于阴道疾病的早期诊断,如有症状的BV。此外,对宏转录组数据的分析结果可以指导创新策略的制定,以调节阴道微生物群的组成和活性,从而恢复和维持最佳的保护环境。
不感兴趣
看过了
取消

 打赏
打赏
不感兴趣
看过了
取消
精彩评论
相关阅读




中国医院排行榜















 010-82736610
010-82736610
 股票代码: 872612
股票代码: 872612



©2012-2023 北京华媒康讯信息技术股份有限公司 All Rights Reserved. 注册地址:北京 联系电话:82736610
 京公网安备 11010802020745号
京公网安备 11010802020745号


