
本研究通过选择性敲除复杂肠道微生物组中的baiH基因,发现baiH影响肠道胆汁酸代谢并以微生物群依赖方式调控宿主炎症。
导读
数百个微生物群基因与宿主生物学/疾病有关。解开微生物群基因对宿主生物学的作用仍然很困难,因为其中许多基因是由非模式肠道共生菌编码的,并且在基因上不具有靶向性。确定微生物基因转移方法和构建其基因编辑工具的一般方法将能够从机制上剖析它们对宿主生理学的影响。研究者开发了一个工具,用于识别跨越五个门的多种非模式微生物的基因转移方法,并且通过在体外以及宿主体内调节微生物衍生的短链脂肪酸和胆汁酸证明了这些基因工具的实用性。在一项原理循证研究中,通过敲除复杂微生物组中负责胆汁酸合成的共生基因,研究者发现该基因在调节结肠炎症中发挥作用。这项技术将使非模式肠道微生物组的基因工程成为可能,并促进微生物群-宿主相互作用的机制剖析。
图文摘要
论文ID
原名:Genetic manipulation of gut microbes enables single-gene interrogation in a complex microbiome
译名:肠道微生物的基因编辑使复杂微生物组中的单基因研究成为可能
期刊:Cell
IF:66.85
发表时间:2022.1
通讯作者:郭春君
通讯作者单位:美国康奈尔大学威尔康奈尔医学院
DOI号:10.1016/j.cell.2021.12.035
实验设计
结果
1.GM工具概述
图1A总结了GM的工作流程。构建这样的基因编辑工具面临三个挑战。首先,以前没有报道过普遍作用于大多数非模式微生物的抗生素标志物。通过评估抗生素耐药性并测试不同的接合供体以将抗生素标记物引入这些肠道分离株中,我们发现一种氯霉素耐药标记在许多转化的微生物中起作用(图1B)。其次,Firmicutes/Clostridia微生物在健康人体肠道中非常丰富,但对这种对宿主生理非常重要的、与宿主相关的细菌群基因编辑在很大程度上仍未得到探索。基因工具的缺乏极大地限制了对Firmicutes/Clostridia基因对宿主生物学影响的机制剖析。通过多参数优化,我们设法确定了多种非模式肠道梭菌微生物的基因转移方法(图1)。第三,在构建工具时,许多分离株的基因组尚未测序,这为大规模建立其靶向基因编辑工具带来了极大障碍。为了克服这一障碍,我们整合了CRISPRi和lacZα转录报告基因或开发靶向细菌16S rRNA基因的遗传策略(图1A)。为实现一致性,如果外源DNA(穿梭或自杀质粒)可以在体外重复引入微生物,我们认为肠道微生物具有基因靶向性。如果在感兴趣的微生物中实现了对其物基因或基因表达的靶向操作,即构建了一种基因编辑工具。
图1. 非模式肠道共生菌的基因编辑(GM)工具概述。
(A) GM工具应用于来自5门、140种的200个人类肠道分离株。该工具确定了88种非模式肠道微生物的基因转移方法,并为其中71种构建了基因编辑工具(表S1)。对于大多数革兰氏阴性肠道微生物而言,通过嵌合16S rRNA策略一步完成其基因转移方法的确定及其基因插入工具的构建。(B)通过GM工具识别的基因靶向微生物16S rRNA序列的系统发育树(按科着色)。(C)本研究中鉴定的基因靶向微生物的详细系统发育信息(另见表S1)。
2.选 择肠道微生物并筛选其培养条件和抗生素耐药性
我们优先考虑在健康人体肠道中占主导地位的厚壁菌门/拟杆菌门微生物,但许多微生物(如Clostridia和Prevotella)没有基因转移方法和易于处理的基因工具。我们通过从各种属/种中选择共生体来使筛选池多样化(表S1)。我们首先确定了支持这些肠道微生物生长的培养条件(图1A;表S1和S3)。接下来,我们针对一系列抗生素对这些微生物进行筛选,以确定以下几点(图1A;表S1):(1) 敏感抗生素的最小抑制浓度(MIC),使其抗性基因可用作选择标记;(2)这些微生物对其有耐药性,但该药物对大肠杆菌有活性,在接合后能抑制大肠杆菌的生长。对于(1),我们确定了抑制几乎所有测试微生物生长的甲砜霉素的MIC(表S1),对于(2),几乎所有的梭菌都对D-环丝氨酸或卡那霉素耐药,而所有的普雷沃氏菌对庆大霉素或D-环丝氨酸耐药(表S1)。
3.确定非模式梭菌基因转移方法的多因子优化
包括不相容的复制起点(rep oris)和抗生素标记、宿主内源性防御系统和低效同源重组(HR)在内的多种原因导致肠道Firmicutes/Clostridia共生菌的遗传学研究相对于其对应的拟杆菌研究不足。因此,在此我们对转化/接合参数进行了多因子优化,以确定先前未转化的肠道Firmicutes/Clostridia共生菌的基因转移条件(图2A)。 首先,由于我们最初将四种最常见梭菌rep oris引入到几种肠道梭菌中(例如C. bolteae)的尝试没有成功,所以我们扩展了Clostridia rep oris库并开发了一种混合接合策略将兼容的rep ori引入非模式肠道梭菌(图2A和S1)。其次,我们利用了一种通用catP标记,这一标记由有效的组成型启动子Ppmtl-catP或Pfdx-cs调控(通过多个非模式梭菌中的启动子库筛选识别;图S1),进而在接合/转化过程中产生抗生素耐药性。当将工具应用于大量非靶向梭菌微生物时,这项工作显著减少了标记物转换的工作负荷(图2A)。第三,我们尝试了不同的方法,包括使用不含大肠杆菌Dcm甲基化酶的“sExpress”接合供体,减少限制性修饰(RM)识别位点、和/或pre-甲基化转化DNA,以减少接合/转化过程中梭菌对宿主防御系统的影响(图2A,STAR方法)。最后,在本研究中优化了其他几个参数,包括接合时间长度、接合供体/受体比例、不同的接合质粒等(图2A;表S1)。
这些协同工作使我们能够以41%的总体成功率确定其基因转移条件(图2A和S1),这表明了开发相关基因编辑系统的可能性。正如预期,需要同时优化多个参数才能成功地将质粒引入以前未转化的梭菌中。例如,将质粒引入S71 C. bartlettii(其具有假定的IV型RM系统),需要一个兼容的Clostridia rep ori,这是一种由强启动子驱动的功能性catP标记,一个不含甲基化质粒DNA并携带R702结合质粒的大肠杆菌“sExpress”供体,以及表S1中详述的接合时间和接合抗生素等其他优化参数的组合。有趣的是,一些梭菌接受不同的rep oris(例如C. bolteae分离株,见表S1),即使它们密切相关,这表明有必要扩展Clostridia rep ori的收集。
图2. 开发非模式肠道共生菌基因编辑工具。
(A) 接合/转化参数的多因子优化示意图,以确定38种大部分未转化的非模式肠道 Firmicutes/Clostridia 的基因转移条件。(B)建立非模式肠道Firmicutes/Clostridia的dCpf1-lacZα平台。菌株的编号对应于表S1中显示的菌株信息。误差棒:SEM。DR,直接重复;G1,指导RNA编码序列1;G2,指导RNA编码序列2;Ter,终止子。(C)非模式梭菌的16S-tron策略示意图。比对梭菌16S rRNA序列以识别Group II intron的保守靶位点。由于RAM(逆转录转座激活标记)的可用性,16S靶向Group II intron(16S-tron)被引入可靶向共生梭菌中。我们鉴定了15个染色体被16S-tron整合的梭菌菌株。(D)Bacteroidia/PrevotellaGM工具的示意图。我们鉴定了31个靶向拟杆菌,其16S rRNA基因已被pGM-NAC2P(或NAC2B)整合。
4.在多种共生梭菌中测试CRISPRi-dCpf1系统
开发梭菌GM工具的下一个关键步骤是确定一种基因编辑工具,该工具可以在大多数梭菌菌株中进行靶向基因编辑。与Cas9启动的切割和dCas9介导的干扰类似,基于CRISPR的系统最近已应用于C. sporogenes和艰难梭菌C. difficile。我们优先选择CRISPRi-dCpf1(失活Cpf1)系统,主要是因为dCpf1不会引发DNA双链断裂,并且dCpf1质粒比Cas9或Cpf1具有更低的毒性和更高的接合效率。相比之下,我们的初步测试发现Cas9/Cpf1的双链切割对许多梭菌来说是致命的,因为它们的HR效率非常低。CRISPRi-dCpf1系统包含一个催化死亡的dCpf1和一个重新用于细菌基因调控的向导RNA(gRNA)。在调控过程中,dCpf1/crRNA复合物与靶基因的模板链结合,阻断转录延伸,从而抑制基因表达。 为了在基因靶向性梭菌中检测CRISPRi-dCpf1,我们将dCpf1和lacZα(作为转录报告基因)与含有9个rep oris的pGM质粒组装在一起(图2B, STAR方法和数据S1B)。选择LacZα是因为其体积小(约300 bp)且在多种梭菌中稳定表达。我们设计了针对lacZα启动子和模板链的双链gRNA(图2B)。我们发现在多种梭菌中dCpf1导致lacZα转录的有效敲除(约3-100倍以上)(图2B和S2;表S1)。一些被测试的梭菌无法接受这组载体,可能是因为该载体体积大(>10 kb)导致接合效率大大降低。总之,我们的数据表明CRISPRi-dCpf1系统调节许多摄取胞外质粒DNA的梭菌中的基因转录。
5.非模式肠道共生菌中靶向细菌16S rRNA基因进而生成靶向基因插入工具的策略
除了CRISPRi,靶向基因插入工具也将有助于研究梭菌菌株基因的分子功能。超过一半的可靶向梭菌都未进行基因组测序。我们想知道,在不了解梭菌基因组序列的情况下,将梭菌普遍保守的DNA序列作为靶标是否可以进行选择性基因插入。然而,高度保守的基因通常是功能上必需的,而这些基因的基因突变可能是致命的。为了找到这一目标,我们研究了已用于评估微生物组多样性和构建细菌系统发育的16S rRNA基因。我们认为16S rRNA基因是一个理想靶标,原因有二:(1)微生物通常具有多个拷贝,破坏其中一个不会致命,并且(2)它在细菌中高度保守。同一组16S靶向载体可以应用于不同的细菌,从而显著节省了测序和克隆的时间和精力。 Group II intron定向突变体系,例如Targetron或Clostron,利用碱基对进行DNA靶识别,引导逆转录转座激活的可选择标记(RAM)位点特异性插入到目标DNA位点。RAM本身被一个自剪接Group I intron中断,只有在剪接出Group I intron并成功插入梭菌染色体后才会产生相应的抗生素耐药性。我们提出,靶向 16S基因的Group II intron可能可以整合到多个梭菌的16S位点。为了检验这一假设,我们比对了16S rRNA基因(来自人类微生物组计划参考基因组),并确定了几个潜在的、高度保守的Group II intron靶位点(图2C)。然后,我们将16S靶向Group II intron (16S-tron)及其兼容的rep oris和抗生素RAM引入18个可靶向梭菌中(图2C;数据S1D)。RAM仅在整合到梭菌菌株后才提供抗生素耐药性。我们发现15个梭菌的染色体被16S-tron插入,表明基于Group II intron的基因插入机制在相应的微生物中起作用(图2C;数据S1D;表S1)。
这些数据表明,该策略可用于开发非模式肠道梭菌的基因插入工具。 然后我们测试了类似的策略是否可以应用于非模式革兰氏阴性肠道共生菌。我们优先考虑了普雷沃氏菌属,因为该属可用的基因工具有限。一般来说,革兰氏阴性菌具有更高效的HR。我们合成了与普雷沃氏菌16S rRNA基因高度同源的嵌合16S(Chi-16S)序列(图2D)。我们将带有自杀质粒的Chi-16S引入21个人类相关的普雷沃氏菌分离株(表S1)。我们发现7个普雷沃氏菌的16S位点被pGM-NAC2P插入(图2D;表S1)。Chi-16S策略还应用于45种 Bacteroides/Parabacteroides微生物和33种来自其他门的肠道相关革兰氏阴性微生物(其中一些具有基因工具),引导我们确定了其中36种菌的基因转移方法(图1C;表S1)。这些数据表明,基于HR的Chi-16S策略有效识别了细菌的基因转移方法,并在非模式普雷沃氏菌和其他门的适合基因操作的肠道相关微生物中生成基因插入工具。
6.构建突变体以调控梭菌基因转录和微生物代谢物生成
为证明本研究中开发的基因工具的实用性,我们选择了一个广泛分布的基因bcat并调控其在11种梭菌和1种双歧杆菌中的表达(图3A)。BCAT蛋白将支链氨基酸脱氨基成酮酸形式(图3A和S3以及表 S1)。引入双链bcat靶向gRNA和dCpf1,与仅含dCpf1的对照组相比,bcat转录在所有具有dCpf1+gRNA的突变体中都受到抑制(图3A)。 随后我们试图利用这些基因编辑工具在体外和宿主定植的情况下调控微生物组衍生代谢物的产生。我们选择了SCFAs中的丙酸盐和丁酸盐,以及支链SCFAs (BSCFAs),因为它们在维持宿主免疫稳态和代谢健康方面发挥着至关重要的作用。我们首先通过分析靶向共生菌的SCFA谱,确定了几种可以产生大量相应代谢物的肠道共生菌。接下来,我们通过靶向相应的代谢基因生成了一系列减少其体外产量的突变体。对于丙酸,我们删除了将甲基丙二酸转化为丙酸的三种拟杆菌属微生物的mmdA基因(图3B和S3;表S1)。针对croA基因,我们使用dCpf1下调其表达量,或使用Group II intron敲除该基因。对于BSCFAs,我们应用dCpf1工具抑制porA在C. sporogenes中的表达。对于我们生成的所有突变体,与对照组相比,相应代谢物的体外产生量显著降低,并且在单定殖无菌小鼠中代谢物的水平也远低于对照(图3D和S3;表S1)。综上,这些数据表明我们可以利用通过GM工具开发的基因工具在体外和宿主定植环境中调控微生物衍生代谢物,这表明它们在微生物群基因及其相应代谢物和相关宿主生物学系统联系间的潜力。
图3. 利用通过GM工具开发的基因编辑工具调节梭菌基因表达和微生物衍生代谢物。
(A)(i) 靶向梭菌中支链氨基酸氨基转移酶bcat的双链gRNA示意图。(ii)使用dCpf1有效抑制了11种梭菌微生物的bcat基因。该面板显示了qPCR测定的三个生物重复的平均基因表达。(B) (i)使用pGM载体敲除拟杆菌mmdA基因的示意图。pMG载体由约1 kb大小的mmdA基因片段组装而成,通过单交叉整合敲除三种拟杆菌微生物的mmdA基因。(ii) 三种拟杆菌ΔmmdA突变体(S25、S27和S31)在体外消耗丙酸盐。衍生细菌培养上清液,并使用LC-MS (EIC: 216.1137)检测丙酸盐生成。拟杆菌ΔmmdA突变体在体内消耗丙酸。无菌Swiss Webster小鼠(每组n=3或4)用S25对照菌株(Con,pGM-NAC2B整合16S)和ΔmmdA突变株(Mut)单定殖。mmdA敲除后宿主中的丙酸盐被耗尽。星号表示p值<0.05 (*)或<0.01(**)。(C) (i)使用dCpf1或Group II intron调节梭菌共生体中丁酸盐产生的示意图。(ii) 三种梭菌微生物S100(dCpf1)、S115(Group II intron)和S117(dCpf1)中的丁酸盐产量(LC-MS定量)显著降低。S117突变体(Mut,dCpf1+gRNAs)单定殖的无菌Swiss Webster小鼠的粪便丁酸盐(LC-MS定量)与对照(Con, 单独dCpf1)相比显著降低。星号表示p值<0.05(*)或<0.01(**)。(D) S107使用CRISPR-dCpf1在体外和体内消耗BSCFAs。(i)使用CRISPR-dCpf1靶向BSCFAs基因porA的示意图。dCpf1 gRNA (G1)靶向porA启动子区域。(ii)与体外对照组(Con,单独dCpf1)相比,突变体(Mut,dCpf1+gRNA)的porA表达(qPCR定量)显著降低。porA抑制突变株单定殖的无菌Swiss Webster小鼠(每组n=4)粪便中的异戊酸少于对照组(单独dCpf1)。星号表示p值<0.05 (*)或<0.01(**)。
7.梭菌特异性胆汁酸7α-脱羟基化的案例研究
我们试图使用这些基因工具来研究一种微生物群基因对宿主生物学的影响。我们选择bai操纵子(CA(胆酸)/CDCA(鹅去氧胆酸)经7α-脱羟基化转化为DCA(脱氧胆酸)/LCA(石胆酸))进行后续研究。促使我们选择此途径的三个原因如下(图4):(1)我们发现了一种可有效将CA(1)/CDCA转换为DCA(3)/LCA的共生Faecalicatena contorta S122 (S122)(图4A和4B)。既往的研究已经逐步阐明了7α-脱羟基化的化学和酶学。然而,研究bai操纵子生物学的一个关键障碍是所有已鉴定的bai编码的梭菌都没有发表的基因转移方法和易于处理的基因工具。(2) DCA/LCA及其衍生物在宿主次级胆汁酸池中占主导地位。(3)两亲性胆汁酸具有抑制肠道病原体生长、调节黏膜免疫、促进肝癌发生等有趣的生物活性。 我们对S122进行测序并鉴定了其对应bai操纵子(图4A)。生物信息学分析揭示了S122及其近亲的三个有趣特征:(1)在两个独立队列中,它们广泛分布于健康人群中(41.30%, 85.98%)。(2)与其他7α-脱羟基梭菌一样,它们的肠道丰度较低(~0.016%),但其bai操纵子在宿主定植条件下转录活跃(图4A)。(3)它们比C. hiranonis或C. scindens更常见、丰富(图S4A和S4B),表明它们在调节肠道7α-脱羟基活性中发挥重要作用。
为了在体内编辑bai通路,我们生成了一个baiH插入突变体(S122 ΩbaiH)(图4B)。baiH基因编码一种氧化还原酶,可将3-oxo-4,5–6,7-didehydro-DCA (2)还原为3-oxo-4,5-dehydro-DCA(图4B)。ΩbaiH突变体在体外消耗DCA并积累中间体(2)和7-oxo CA(图4B)。出乎意料的是,我们试图有效地将S122单独定殖于无菌小鼠以诱导体内DCA产生,但这一尝试失败了。相反,我们发现S122与S25可以稳定地共定殖于无菌小鼠,并且敲除baiH消除了肠道7α-脱羟基活性:对照组积累了约12 pmol/mg DCA(图4C和4D),而突变体消耗了DCA但积累7-oxo-CA(图4D)。此外,S122可以稳定地与本研究中鉴定的其他55种基因靶向微生物共定殖无菌小鼠(图S4E)。这两种情况下S122的相对丰度都很低(图4C),但可以检测到从CA到DCA的强力转化,这表明在宿主体内S122是一种高活性的7α-脱羟基细菌。
图4. 在定菌小鼠中敲除baiH。
(A)胆汁酸7α-脱羟基化中假定的S122 bai操纵子的定位。突变基因baiH(通过Group II intron)以红色突出显示。S122 bai操纵子在宿主中定植时活跃转录,并给出S122 bai操纵子宏转录组学分析的三个代表性结果。(B)胆汁酸7α-脱羟基的生物合成体系。baiH编码一种氧化还原酶,可还原中间体3-oxo-4,5–6,7-didehydro-DCA的6,7-烯烃键(2,EIC:385.2384)。S122 ΩbaiH突变体在体外积聚预测中间体并且不再将CA转化为DCA。中间体(2)的结构通过将其保留时间和精确质量与已发表的文献进行比较来确定。(C)S25+S122对照(Con)或ΩbaiH突变株(Mut)(通过Group II intron)共同定殖于无菌C57BL/6J小鼠(每组n=3或4)。通过16S rRNA测序评估对照组和突变组中S122的相对丰度,两者具有可比性。(D)使用Group II intron敲除baiH会破坏小鼠肠道7α-去羟化活性,并改变肠道胆汁酸池。使用LC-MS量化粪便CA、DCA和7-oxo CA。(C)和(D)中的数据使用非配对双尾t检验进行分析。星号表示p值<0.05 (*)或<0.01(**)。
8.baiH基因显著影响宿主胆汁酸池和微生物群组成
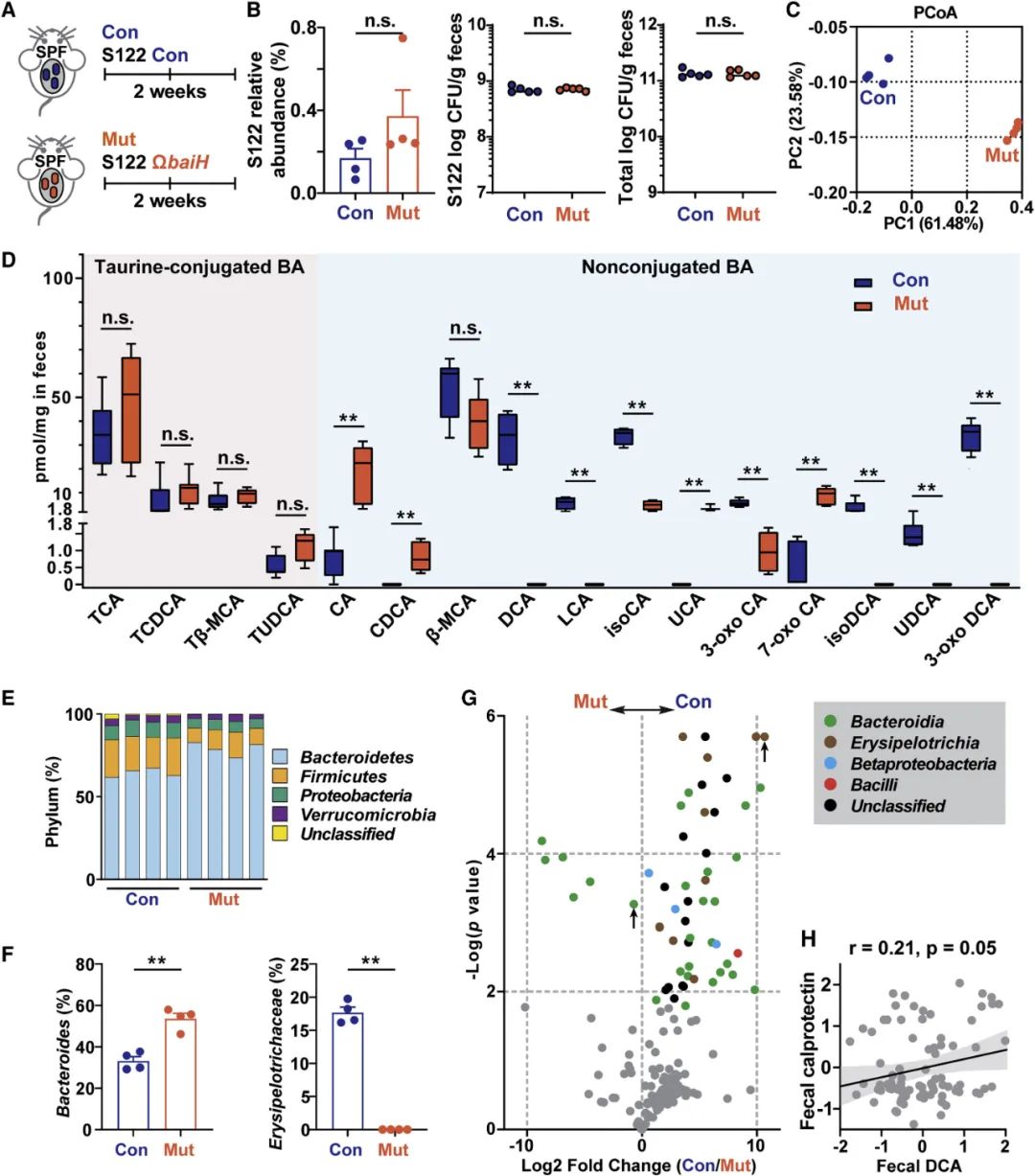
这一发现促使我们敲除复杂的微生物群中的baiH,如SPF小鼠。编辑复杂微生物组中的微生物群基因可以直接读出它们对微生物组成的影响,这对于解释其对宿主生物学的影响至关重要。与无菌小鼠不同,SPF小鼠的胃肠道已拥有一个具有强大的7α-脱羟基活性的复杂微生物组,为S122的定殖留下了有限的生态位。为了克服这一挑战,我们在对照组和ΩbaiH突变组进行甲砜霉素抗性基因标记。我们在小鼠饮用水中添加了极低浓度的甲砜霉素(15 μg/ml)和红霉素(10 μg/ml),原因有二:(1)促进对这两种抗生素耐药的标记S122菌株的定殖,以及(2)消除现有的bai编码梭菌所赋予的背景7α-脱羟基活性。该策略使S122对照组和ΩbaiH突变体在4周内以相同水平稳定定殖于SPF小鼠中,且总细菌负荷相当(图5A和5B)。因为添加的抗生素在粪便中积聚最少(甲砜霉素约为5 pmol/mg,红霉素检测不到),与SPF小鼠相比,它们不会降低总细菌负荷。在这种实验环境下它们对肠道微生物组的影响也受到控制。
为检测baiH缺失是否影响肠道胆汁酸组成和微生物组,我们对小鼠粪便样本进行代谢组学和16S rRNA测序分析(图5和S5)。主坐标分析(PCoA)表明粪便样本按基因型聚集(图5C)。我们从这些数据中得出两个观察结果:首先,对照组和ΩbaiH定殖小鼠具有不同的肠道胆汁酸池。两组的TCA和TCDCA等结合胆汁酸水平相似(图5D),表明baiH缺失不会显著改变微生物组胆汁盐水解活性。DCA及其衍生物(如isoDCA和3-oxo DCA)在对照组中积累的水平与宿主生理水平相当。与此相反,突变组的CA及其衍生物水平更高,包括7-oxo CA和熊胆酸(UCA)(图5D)。其次,敲除baiH会改变宿主肠道微生物组的组成。对照组和突变组都具有高度复杂的粪便微生物群,并且它们的整体门水平组成保持不变(图5E)。对照组的拟杆菌门丰度较低,变形菌门的丰度较高,丹毒丝菌科Erysipelotrichaceae丰度显著升高(图5F)。这种组成变化与肠道炎症恶化有关。各组之间共有56个操作分类单元(OTU)差异丰富,它们主要是拟杆菌、β变形菌Betaproteobacteria和丹毒丝菌(图5G)。值得注意的是,对照组的丹毒丝菌明显更多,这些菌体具有高IgA涂层,并且与加重的结肠炎症相关(图5F和5G)。与我们在SPF小鼠中的发现一致,在非IBD人群中,较高的粪便DCA与丹毒丝菌科丰度和粪便中的粪便钙卫蛋白(肠道炎症水平的标志物)呈正相关(图5H)。然而,在溃疡性结肠炎或克罗恩病患者中没有观察到这种相关性,这些患者的肠道微生物群通常发生结构改变,并且由于过度的免疫反应而导致肠道7α-脱羟基活性被破坏。这些数据表明bai操纵子在人体肠道微生物群和肠道炎症的发生中具有潜在调控作用。
图5. 在复杂微生物群的背景下敲除baiH会影响宿主胆汁酸池和肠道微生物组。
(A) 给予低剂量抗生素(15 μg/ml甲砜霉素和10 μg/ml红霉素)的SPF C57BL/6J小鼠(每组n=4或5)被基因标记的S122对照(Con)或ΩbaiH突变株(Mut)定殖。(B)通过16S rRNA测序评估对照组和突变组中S122的相对丰度,两者具有可比性。S122对照(Con)和ΩbaiH突变株(Mut)以大致相同的水平稳定地定殖于SPF小鼠,并且总细菌负荷相当。(C)对照组和ΩbaiH突变组小鼠粪便微生物组的主坐标分析(PCoA)。(D)对照组(Con)和ΩbaiH突变株(Mut)定殖的SPF小鼠粪便胆汁酸(BA)成分的靶向代谢组学分析(LC-MS定量)。(E)对照组和ΩbaiH突变组小鼠肠道微生物群中门水平的相对丰度。(F)对照组和ΩbaiH突变组小鼠粪便微生物组中炎症相关肠道微生物类群的相对丰度。(G) 16S rRNA基因测序计算差异细菌OTU丰度的火山图。着色并绘制具有显著差异的OTUs (n=56, FDR<0.05)。相对丰度高 (>10%)的拟杆菌OTU和丹毒丝菌科OTU用向上箭头标记。(H)非IBD人群中肠道7α-脱羟基活性与粪便钙卫蛋白水平弱相关。
9.评估baiH对肠道炎症的影响
因为敲除baiH会将肠道微生物组转变为弱促炎状态,我们在DSS诱导的小鼠结肠炎模型中评估了baiH是否调控肠道炎症反应。对照组和ΩbaiH突变株定殖的SPF小鼠饮用含有DSS的水(图6A和6F)。我们发现S122对照组和ΩbaiH突变株均稳定地定殖于小鼠,而对照组在DSS处理期间仍然具有显著较高水平的丹毒丝菌科菌株。随着结肠炎症的进展,baiH的确在肠道炎症中发挥调控作用:对照组体重减轻更多,炎症更加严重,表现为结肠病理学增强、结肠长度缩短、粪便lipocalin-2水平升高、便血评分增加和炎症基因上调(图6B-6E)。有趣的是,相同的DSS治疗成功地引发了S122对照组或ΩbaiH突变株和S25共定殖的无菌C57BL/6J小鼠中的炎症反应,但敲除baiH对肠道炎症没有显著影响(图6G–6J)。综上所述,这些数据表明baiH介导的炎症反应依赖于微生物群,而复杂微生物群中的baiH敲除会重塑宿主胆汁酸谱,并预先将肠道微生物群组成调整为更具保护性的状态来对抗DSS诱导的结肠炎。更重要的是,通过微生物组遗传学、代谢组学和结肠炎小鼠模型的结合,我们展示了低肠道丰度的单个共生体基因如何通过重塑胆汁酸代谢和肠道微生物群生态系统进而显著影响宿主生物学。
图6. baiH在复杂肠道微生物群的背景下调控肠道炎症。
(A、F)采用S122对照(Con)和ΩbaiH突变株(Mut)定殖SPF或定菌小鼠建立DSS诱导的小鼠结肠炎模型。在给予DSS之前,用对照或突变株定植小鼠至少2周,给予SPF小鼠2.5% DSS(在添加15 μg/ml甲砜霉素和10 μg/ml红霉素的水中)8天,同时给予无菌小鼠2.0% DSS(在添加15 μg/ml甲砜霉素的水中)7天。通过体重减轻(B和G)、远端结肠(C和H)H&E染色、结肠缩短、组织病理学评分(D和I)、粪便lipocalin-2和每日便血评分来监测疾病状态(E和J)。数据显示为平均值±SEM。星号表示p值<0.05 (*)、<0.01(**)或 <0.001(***)。
10.baiH介导的菌群组成变化加重了DSS诱导的定菌小鼠结肠炎
受baiH在复杂微生物组中介导结肠炎症这一发现的启发,我们进一步研究了baiH缺失诱导的微生物群组成变化(图5F)是否与DSS处理下SPF小鼠的不同肠道炎症反应有关。首先,我们确定丹毒丝菌科菌株的扩张是否依赖于baiH。事实上,与拟杆菌菌株相比,丹毒丝菌科分离株对高浓度DCA和3-oxo DCA的抵抗力更强(图7A)。在体外制备的含10个成员的合成菌群中,在baiH及其产物DCA存在条件下,丹毒丝菌科类群同样扩张(图7B和7C)。
随后我们调查了baiH是否会推动体内丹毒丝菌科类群的扩张,以及这种微生物群组成的变化是否会影响DSS结肠炎模型中的结肠炎症反应。我们用相同的含10个成员的合成菌群(S122对照或ΩbaiH突变株联合7种丹毒丝菌科和2种拟杆菌菌株,图7B和7D)对两组无菌C57BL/6J小鼠定殖,并在定殖后两周给予DSS处理(图7D)。正如预期,在宿主定植的背景下baiH推动了丹毒丝菌科的扩张(图7E)。在这种限菌环境中对照组的结肠炎症加重,通过严重的体重下降(图7F)、结肠病理学增强(图7G)、结肠长度缩短(图7H)、粪便lipocalin-2水平升高(图7I)和便血评分的升高(图7J)来评估炎症。在无菌C57BL/6J小鼠中相同的DSS处理同样诱导了强烈的炎症反应,该小鼠中S122对照或ΩbaiH突变株与2个拟杆菌共同定殖(3个成员)。然而,在这种限菌环境中敲除baiH对肠道炎症没有显著影响。S122对照和ΩbaiH突变株在两种限菌环境(10个成员vs. 3个成员)中以相当的水平定殖小鼠,且它们的粪便胆汁酸谱相似,这表明在10个成员联合定殖小鼠中观察到的不同肠道炎症反应更可能是由于baiH驱动的丹毒丝菌科的扩张。总之,这些数据表明baiH介导的微生物群组成变化可能会加剧DSS诱导的定菌小鼠结肠炎,并且在SPF小鼠中观察到的类似变化可能与复杂微生物群中baiH敲除诱导的不同肠道炎症反应有关(图5F)。值得注意的是,合成菌群的成员是根据高度多样化的微生物组中敲除baiH获得的信息来选择的,这证明了在复杂微生物群背景下研究微生物群基因功能的有用性和必要性。
图7. baiH介导的微生物群组成变化加重DSS诱导的定菌小鼠结肠炎。
(A) 在500 μM DCA、500 μM 3-oxo DCA或DMSO对照存在下两种拟杆菌(Bac)微生物和七种丹毒丝菌科(Ery)微生物的生长曲线。丹毒丝菌科微生物对DCA和3-oxo DCA的抵抗力比拟杆菌微生物强。(B) baiH基因驱动的丹毒丝菌科微生物在由两个拟杆菌(Bac)和七个丹毒丝菌科微生物(Ery)与S122对照或ΩbaiH菌株组成的体外菌群中扩增。补充500 μM CA作为bai途径的底物。通过qPCR评估丹毒丝菌科的相对倍数变化。(C) 在由两种拟杆菌(Bac)微生物和七种丹毒丝菌科(Ery)微生物组成的体外菌群中,DCA推动丹毒丝菌科微生物的扩增。分别补充0、250和500 μM的DCA。通过qPCR评估丹毒丝菌科的相对倍数变化。(D) 将DSS诱导的小鼠结肠炎模型应用于由基因标记S122对照(Con)或ΩbaiH突变株(Mut)以及拟杆菌(Bac)微生物和七种丹毒丝菌科(Ery)微生物组成的合成菌群定殖的无菌小鼠,分别在(A)、(B)和(C)中检测。用对照或突变株定殖小鼠至少两周,然后2.5% DSS给药8天。(E)在DSS处理前和处理期间,通过qPCR评估baiH基因驱动的丹毒丝菌科微生物的扩张。通过体重减轻(F)、远端结肠(G)的H&E染色、结肠缩短(H)、粪便lipocalin-2(I)和每日便血评分(J)监测疾病状态。数据显示为平均值±SEM。星号表示p值<0.05(*)、<0.01(**)或 <0.001(***)。
讨论
本研究中的GM工具用于识别可基因靶向的肠道微生物并构建其基因编辑工具,与其他基于微生物组的基因工具集协同作用,增加额外的肠道微生物组工程方法,促进更好地理解微生物-宿主相互作用。GM以体外、高效、高通量的方式进行筛选,并显著扩展了肠道微生物组的可操作范围。此外,通过整合CRISPR体系和靶向细菌16S rRNA基因(例如16S-tron和Chi-16S),我们在基因组序列未知的情况下通过“筛选”的方法在多种系统发育多样性的微生物中构建了基因编辑工具(表S1)。我们提供了这些非模式肠道共生菌的基因转移方法及其基因工具,为科学界中进一步研究其基因分子功能或剖析它们与宿主相互作用背后的分子机制提供了资源。 研究微生物组基因最精确的方法是在复杂微生物群的背景下对其进行编辑,并比较仅在该基因中存在差异的肠道微生物组组成和宿主表型。这种方法类似于宿主条件基因敲除。虽然这种技术已被广泛应用于敲除特定组织中的宿主基因,但目前还没有可用于肠道微生物组研究的类似工具集。
本研究通过选择性敲除复杂肠道微生物组中的baiH基因,发现baiH影响肠道胆汁酸代谢并以微生物群依赖方式调控宿主炎症。更有趣的是,在SPF小鼠中观察到的微生物群组成变化引导下,我们进一步在无菌小鼠中建立了一个合成菌群,并证明baiH驱动的微生物群组成变化可以影响宿主结肠炎症反应。该技术和方法可用于研究其他微生物群基因的生物学功能和相关代谢途径,并揭示肠道微生物组组成变化调控的宿主生物学。本研究提出的GM工具将极大地促进微生物群基因与疾病因果关联的功能研究或微生物-宿主相互作用的分子机制研究。人类微生物组是“泛基因组”的重要组成部分,其生物学功能可能通过基因编辑调控。虽然目前没有对整个肠道微生物组进行基因编辑的技术,但我们相信GM工具是一种高效且可推广的用于识别可基因靶向非模式肠道共生菌并构建其基因工具的方法,这将是建立人类肠道微生物组基因编辑技术的第一步。
编译:微科盟蔚蓝,编辑:微科盟居居、江舜尧。
微科盟原创微文,欢迎转发转载,转载须注明来源《微生态》。
不感兴趣
看过了
取消

人点赞

人收藏

打赏
不感兴趣
看过了
取消
©2012-2023 北京华媒康讯信息技术股份有限公司 All Rights Reserved. 注册地址:北京 联系电话:010-82736610
广播电视节目制作经营许可证 —(京)字第 17437号 京海食药监械经营备20200522号
京ICP备12011723号 京ICP证150092号
 京公网安备 11010802020745号
工商备案公示信息
互联网药品信息服务资格证书((京)-非经营性-2020-0015)
京公网安备 11010802020745号
工商备案公示信息
互联网药品信息服务资格证书((京)-非经营性-2020-0015)






您已认证成功,可享专属会员优惠,买1年送3个月!
开通会员,资料、课程、直播、报告等海量内容免费看!

打赏金额
认可我就打赏我~
1元 5元 10元 20元 50元 其它
打赏作者
认可我就打赏我~
扫描二维码
立即打赏给Ta吧!
温馨提示:仅支持微信支付!
已收到您的咨询诉求 我们会尽快联系您








































 010-82736610
010-82736610
 股票代码: 872612
股票代码: 872612




