最近利用测序技术和分析方法发展的研究强调了结构变异在人类和其他动物、各种植物以及最近的细菌中的普遍性和功能重要性。与单核苷酸多态性(SNPs)相比,结构变异在基因组中的发生率较低,但在真核生物中有更高的机会影响基因功能。
编译:微科盟小木,编辑:微科盟小编、江舜尧。
微科盟原创微文,欢迎转发转载,转载须注明来源《微生态》公众号。
导读
深入分析肠道微生物组的遗传变异对于理解其功能和对宿主健康和疾病的影响非常必要。本研究通过利用纳米孔技术(ONT)提供的长读长优势,表征了来自健康人体的数百个肠道微生物组中结构变异(SVs)的精细遗传变异。ONT长读长极大地提高了宏基因组组装的质量,能够可靠地检测大量扩展的结构变异类型(特别是包括大型插入和倒置)。我们发现,SVs在个体间高度不同,在个体内稳定,代表肠道微生物组指纹,这些指纹塑造了物种内功能的菌株水平差异,使与代谢物和宿主表型(如血糖)的关联复杂化。综上,本研究强调将ONT reads纳入宏基因组分析扩大了遗传变异的检测范围,使分析肠道微生物组中的菌株水平变异及其与代谢组的复杂相关性成为可能。
论文ID
原名:Short- and long-read metagenomics expand individualized structural variations in gut microbiomes
译名:短读长和长读长宏基因组学扩展了肠道微生物组的个体化结构变异
期刊:Nature Communications
IF:14.919
发表时间:2022.6.8
通讯作者:王军&宋默识
通讯作者单位:中国科学院微生物研究所&中国科学院动物研究所
DOI号:10.1038/s41467-022-30857-9
实验设计
结果
1 混合测序提高了人体肠道宏基因组组装的质量
我们首先建立了一个包含ONT和Illumina reads的混合管道,并实现宏基因组组装和后续数据分析。利用ZymoBIOMICS微生物群落(包含8株细菌等摩尔的DNA)产生的ONT和Illumina reads,该管道实现了高完整性(94.54-99.75%)、低污染率(0-6.97%)和>97.6%的平均核苷酸同源性(ANI)用于重建细菌基因组(方法,补充图1a-e)。相比之下,仅使用Illumina reads会导致更短的contigs,分箱基因组的数量及其完整性没有可检测到的变化;使用旨在生成循环基因组的方法分析ONT reads,基因组完整性总体低于93%,同时分箱基因组数量减少(补充图1b和补充数据1)。我们进一步检查了ONT reads是否会给contigs引入更多错误,并减少基因组中的开放阅读框(ORF)数量(即编码密度),发现与仅使用Illumina组装相比,混合组装的编码密度没有明显降低,而仅使用ONT reads导致编码密度显著降低(补充图1c)。
然后,我们将混合组装策略应用于两组人体肠道微生物组数据:100名健康个体组成的横断面队列(补充数据2)和10名健康个体组成的时间序列队列(补充数据3),每个人连续收集10个粪便样本。结果表明,我们的混合组装方法大大提高了来自两个队列的200个粪便样本的宏基因组contigs的质量。每个样本平均使用1.4×10 6 个ONT reads(平均长度为5683 bp)和5.6×10 7 个Illumina 150 bp对端reads,我们的管道平均组装了7.1×10 7 个contigs,总计7.6×10 10 bp(补充图1f和表1)。
总体而言,与仅使用Illumina reads获得的组装相比,混合组装的contigs减少了17.3%,总组装序列增加了5.1%(分别为8.5×10 7 contigs和7.2×10 10 bp,表1)。值得注意的是,与单独的短读长组装(2962 bp)相比,混合组装(9283 bp)的平均N50值增加了三倍多。 然后,我们将混合组装获得的contigs分箱成代表单个细菌物种的宏基因组组装基因组(MAGs),共得到9612个MAGs(每个样本20-83个MAGs),平均N50为117 kb;去除冗余的MAGs后,仍有692个MAGs(即属于同一细菌物种,图1b、c和表1),其中623个对应于UHGG数据库中可用的基因组分箱,其中208个在我们的混合组装结果中具有更高的质量;总共67个基因组分箱中的其余部分是新基因组。
去重复后减少了两个MAGs,因为我们的队列和UHGG集合中一些以前没有聚集在一起(dRep13 v2.2.4)的相对接近的MAGs现在与更高版本的dRep聚集在一起(我们使用v2.6.2)。在全面性方面,159个(22.97%)非冗余MAGs包含所有3种类型的rRNA序列(23S、16S和5S),448个(64.74%)MAGs包含至少1种类型的rRNA。相比之下,仅Illumina组装产生的非冗余MAGs减少了11%(616个),N50值约为平均N50值(65 kb)的一半,其中只有9个(1.46%)MAGs具有所有三种rRNA序列,只有258个(41.88%)具有至少一种rRNA序列。
在所有样本中,最常见的MAG是 Fusicatenibacter saccharivorans (存在于172个样本中),其次是 Anaerostipes hadrus (150个样本)和 Agathobacter rectalis (148个样本),在10个以上的样本中有189个物种以MAGs的形式存在(补充数据4和补充图2)。
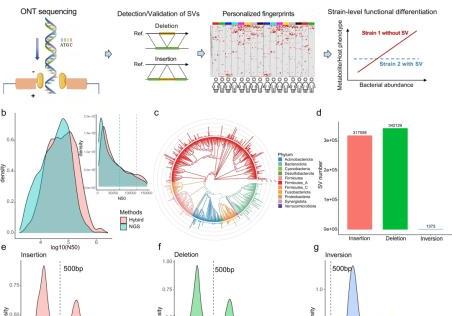
图1 ONT reads改进的宏基因组组装,支持结构变异(SVs)检测和验证
a本研究的工作流程示意图。首先,利用来自ONT的长读长,我们改进了数百个肠道微生物组中的宏基因组组装,使大型SVs的检测成为可能,特别是包括插入和倒置,这是高度个性化的肠道微生物特征,并使其与代谢物或宿主健康指标的相关性复杂化。b仅采用Illumina方法(NGS)和混合组装(Hybrid)的contig长度分布,完整分布如主图中的对数刻度所示;部分详细分布显示在右上图中,包括虚线,显示了在仅采用Illumina方法(NGS)和混合组装(Hybrid)中,binned宏基因组组装基因组(MAGs)的平均N50值。c采用混合组装对200个肠道微生物组样本中的692个MAGs进行系统发育分布分析。
颜色表示主要的肠道微生物门,外圆的线表示200个样本中MAGs(物种)的出现次数。d在我们队列中发现的存在于10个以上个体中的189个MAGs(物种)的插入、缺失和倒置总数,每个MAGs(物种)有一个代表性MAGs,其余MAGs与该代表性MAGs进行比较(见方法和结果)。e-g显示发现的插入、缺失和倒置的长度分布,特别是大型SVs(>500 bp),约占插入和缺失以及所有倒置的50%。
表1 每个样本的混合组装和仅采用Illumina组装的关键参数汇总
对于每个参数,显示平均值和范围。 a表示混合组装中使用的ONT reads数量。
2 扩大肠道微生物组结构变异的检测范围
改进的N50>110 kb的宏基因组组装为扩大人类肠道微生物组遗传变异的研究范围提供了机会。特别是,考虑到缺失仍然是基于参考基因组映射在肠道微生物组中研究的主要结构变异类型,而插入和倒置,尤其是短读长无法覆盖的大片段插入和倒置仍然难以捉摸。更长的ONT reads改进了宏基因组组装,并能够发现包括插入/倒置在内的扩展SVs,同时提供了在整个队列的reads水平对大型SVs进行直接验证的机会。
在我们的横断面队列中,我们通过比较MAGs可靠地检测到多种类型的结构变异。对于存在于10个以上个体中的189种细菌,我们使用dRep v2.6.213评估的得分最高的MAGs作为比较来自其他样本的同种细菌MAGs的参考。这确定了总共317,558个插入、342129个缺失和1373个倒置(图1d)。 值得注意的是,大于500 bp的SVs在每种SV类型中占很大比例,包括170,329(53.63%)个插入,184,037(53.80%)个缺失,以及所有1373个倒置(图1e-g)。有趣的是,我们观察到插入和缺失分布有两个峰,为此我们假设这两个峰是原核基因组中不同生物学过程的结果,特别是在转座子/原噬菌体和其他移动元件活动方面。因此,我们随机选取两个峰内的SVs(分别在140-160 bp和1050-1150 bp内)进行分析,并基于mMGE数据库使用blastn预测原噬菌体和染色体外移动遗传元件(eMGEs)。结果表明,两个峰内的SVs之间存在显著差异,并且短SVs中的移动元件显著高于长SVs:短SVs vs. 长SVs中的原噬菌体:缺失(p=2.82e-06)、插入(p=2.93e-05);短SVs vs. 长SVs中的eMGEs:缺失(p=4.385e-07)、插入(p=0.0005129,均采用Wilcox检验)。因此,我们推断与较长的SV相比,短SV更可能是噬菌体整合和其他移动元件的结果。然而,由于并非所有SVs都具有可检测的移动元件,这仅提供了部分且合理的解释;我们假设其他SVs是复制错误或重组事件的结果,但从专注于细菌中SVs的有限研究中无法获得机制验证。
我们基于重新映射到参考MAG或包含SV的MAG,通过识别可以直接覆盖SV及其侧翼区域的ONT reads来评估检测到的SV的可靠性。人工检测最终证实,在随机选择的SVs集合中,>97%的SVs得到多个ONT reads的支持,从而可靠地验证了具有覆盖相应基因组区域的单分子reads的特定SVs的存在(图2a和补充图3);映射结果同时表明,同一个体内同种细菌基因组的SVs异质性较低。 在我们的SV数据集中,一个明显的趋势是细菌基因组中SV的频率在分类组之间是不均匀的。在物种(MAGs)水平上,我们的数据显示,发现的SV总数与MAGs的数量成正比,也与所有样本的基因组大小成正比。因此,为了解释SV分布的不均匀,我们的研究结果强调,不同分类组之间的任何比较都应包括基于成对比较的平均值,并应校正基因组大小(补充图4)。
对给定MAG和参考MAG之间的平均SV数量的分析(在分类组中标准化为每1 Mb基因组)表明,在门水平上,紧随SVs高度多样化的厚壁菌门(SVs中位数为20.4)之后,包含 Akkermensia 的细菌门Verrucomicrobia的SVs数量位居第二(中位数19.5,图2b,c),而脱硫菌门(Desulfobacteroita)和变形菌门(Proteobacteria)的SVs数量最低(中位数分别为8.6和11.5,图2b)。疣微菌门(Verrucomicrobia)只包含一个已确定的物种,即 Akkermensia muciniphila ,目前正在广泛研究其代谢调节作用。在本研究中,它们个体间的高遗传多样性和由此产生的更大的泛基因组要求在解决转化研究中的菌株水平差异时需要特别注意。
图2 人体肠道微生物组结构变异(SVs)的验证和表征
a使用长ONT reads直接验证结构变异(SVs)的示意图。以上部宏基因组组装基因组(MAGs)为参考,识别下部MAG(属于同一细菌物种,来自不同样本)的缺失(左)和插入(右),并将来自不同样本的长reads与代表性序列进行映射导致reads直接覆盖缺失(左)或插入(右)和侧翼区域,从而验证这些SVs在read水平的存在。b,c不同分类组中主要SVs类型(插入和缺失)数量的门水平和科水平分布,根据相应的基因组大小(每1 M基因组的SVs)进行校正。SV数量在不同的门和细菌科中具有很高的变异性(见结果)。d比较用于SV检测的所有189个MAGs之间每1 Mb基因组中SV的平均数量,以及来自三个最常见的同一物种的单个MAGs,在横断面队列中的不同个体之间(每个箱线图的左侧)以及时间序列数据中来自同一个体的不同样本(每个箱线图的右侧)。在所有四种情况下,个体间SVs数量均显著高于个体内SVs数量(双侧Wilcoxon检验,分别为n=4093、83、91、63,所有P<2e-10),表明SVs可以作为人体肠道微生物组的指纹来区分不同的个体。
3 SVs作为肠道微生物组的高度个性化特征具有功能信息性
对ONT-read-informed SV数据集的分析有力地支持了SV可以定义信息丰富、个性化的肠道微生物组特征的想法。同时我们在横断面队列数据中发现了高的个体间变异性,而在我们的时间序列数据中发现了较低的个体内变异性和时间变异性。对我们SV发现研究中使用的189个MAGs分析显示,在来自不同个体的MAG之间发现每Mb基因组16.7个SVs(中位数),而在十个连续的时间点上,单个个体中每个MAG的中位数为0个SV(Wilcox检验p<2.2e-16,图2d)。
因此,SVs可靠地区分了不同个体之间的细菌种类和肠道微生物组。需要强调的是,我们的研究结果反映了“LifeLines”队列的最新发现,即结构变异包括可以区分个体特异性细菌物种的指纹。此外,除了先前专注于缺失的研究报告的SV指纹(由于基于短读长组装的限制)外,我们的数据集显示几乎相同数量的插入与此类缺失同时存在。我们数据集的时间分辨样本也使我们能够补充这些关于个体特异性肠道微生物组特征的发现:在大约10天内(图2d和补充图5),同一物种的基因组结构保持稳定,这表明在LifeLines队列中观察到的菌株分化/替换可能是SV逐渐积累的结果。 由于基因组中的SVs会引起基因组中的断裂点,从而可能会影响基因的功能,我们研究了包含此类断裂点的参考MAGs中基因的功能分布。
与参考MAGs相比,我们观察到SV的数量和类型在个体间存在较大的差异,因此很难从SV的功能来研究每个个体和细菌基因组;此外,每个个体和每个MAG中SVs数量相对较低(16.7/Mb)阻碍了信息富集分析,因此我们在群体规模上对SVs相关基因功能进行了功能富集分析。使用KEGG通路并对照参考MAG中预测的所有基因的基线,富集分析显示共有267条插入和缺失的富集通路;倒置没有显著富集的通路,可能是因为它们的数量比插入/缺失的数量要少。
在受影响最大的30条通路中,有19条与代谢相关的通路(根据富集程度排序),如聚糖降解、鞘脂代谢和各种碳水化合物代谢(图3a和补充数据5)。与环境信息处理相关的通路也有富集,如磷酸转移酶系统(PTS)、ABC转运体和双系统转运系统,其基因与细菌毒素的产生和赋予抗生素耐药性有关(图3a和补充数据5)。有趣的是,富集结果与先前研究以色列和荷兰人群中SV影响基因的报告中的结果一致,表明人类肠道微生物SV影响基因的潜在普遍特征。
图3 人体肠道微生物组结构变异(SVs)的功能相关性
a基于KEGG的SV影响基因功能富集前30类,代谢相关通路占19个;p值来自Fisher检验。b SVs影响肠道细菌-粪便代谢物相关性,上图显示显著相关性(Spearman相关性,FDR<0.1),SVs的存在消除了显著相关性(下图,所有p>0.05),并且没有SVs的亚群保持显著性(中图,所有p< 0.05)。颜色表示不同的细菌种类。c SV影响的细菌-代谢物相关对概述(Spearman)。颜色和条形表示不同的细菌和代谢物。d Fusicatenibacter saccharivorans与新海藻糖显著相关(NSV, n=100),K03655基因内的SVs导致相关性不显著(SV1, n=52;ρ(rho)=0.032, p=0.82),与其他亚群/菌株不同(SV0, n=48;ρ=0.45, p=0.0015, FDR=0.099)。e A. rectalis和1-磷酸果糖(F1P)之间的相关性类似,K01193处的SVs导致菌株水平差异。f, g粪便新海藻糖和F1P浓度与血糖显著相关(n=100)。h对于F. saccharivorans,K03655基因内的SVs与血糖的相关性不显著(SV1, n=52;ρ=0.20, p=0.15),无SVs的亚群与血糖显著相关(SV0, n=48;ρ=0.54, p=8.6e-5, FDR=0.001)。i同样,对于A. rectalis,K01193基因内的SVs也会导致与血糖相关的菌株水平差异(SV0, p=0.05, n=46;SV1组n=54)。ρ(rho)表示spearman相关系数,p值使用Benjamini-Hochberg程序调整以进行FDR控制。(d-i)中的阴影表示95%置信区间(CI),d-i中的颜色表示不同的组。NSV:所有样本;SV0:细菌基因中不含SV的亚群;SV1:细菌基因中存在SV的亚群。
4 SVs使细菌与代谢物和宿主表型的联系复杂化
为了研究SVs的功能影响,特别是对微生物代谢的影响,我们对横断面队列的粪便、血清和尿液样本进行了代谢组分析。基于横断面队列中不同样本的代谢组分析表明,SVs使细菌种类和代谢物之间的相关性复杂化,导致同一种细菌内菌株水平的功能差异与代谢物显著相关。即SVs导致了基因功能的潜在破坏,并且在包含SVs的亚群中消除了细菌丰度和代谢物之间的显著相关性;相比之下,没有SV的亚群保持了显著的相关性。
在这个更精细的分析中,我们发现在11种与粪便、血清或尿液代谢组中的代谢物有显著相关性(FDR<0.1)的细菌中,共有889个SV影响基因使细菌-代谢物相关性复杂化(图3b、c、补充图6和补充数据6)。我们发现,其中753对涉及70个SVs,与74种粪便代谢物相关(458),134对涉及31个SVs,与66种尿液代谢物相关(396),2对涉及2个SVs,与2种血清代谢物相关。在这些结果中,我们对插入的发现和包含表明SVs的扩展增加了发现可能使细菌-代谢物相关性复杂化的SVs的能力。例如,先前在以色列人群中发现肌醇浓度与 A. hadrus 之间的相关性被细菌基因组的缺失所混淆。相比之下,我们的研究发现,在 Bacteroides uniformis 基因组中的基因位点(注释为K02014,铁复合物外膜受体蛋白)发生的插入和缺失均导致该细菌物种的相对丰度与尿液中肌醇浓度之间的显著相关性消失(补充图7)。 在细菌种类和代谢物之间的SV混杂关联中,我们发现当忽略SVs和其他细菌时, A. rectalis 与粪便1-磷酸果糖(F1P)显著相关(Spearman ρ=0.28, p=0.0053, FDR=0.035,图3e)。对12个受SVs影响的基因状态的进一步分析使 F. saccharivorans 与粪便样本中新海藻糖浓度之间的相关性复杂化(图3d,和补充数据6)。
同样,33个受SVs影响的基因表明,含有SVs的细菌亚群(菌株)不再与F1P显著相关(如 A. rectalis 中的K01193, ρ=0.18, p=0.2, FDR=0.56,图3e)。在代谢物和SV影响基因中,我们发现了4个受SV影响的代谢物,受SV影响的基因共有11个被映射到4个KEGG通路,其中受SV影响的基因和代谢物都参与其中,这强烈表明SV通过影响相关基因的功能来塑造细菌-代谢物的相关性(补充数据7和补充数据8)。例如,SV影响基因混淆了细菌与新海藻糖的关联,包括K01208(环麦芽糖糊精酶)和K05349 (β-葡萄糖苷酶),它们都属于KEGG途径淀粉和蔗糖代谢(地图00500)(补充图8和补充数据7)。总之,我们的数据表明,SVs能够在肠道微生物物种的代谢功能中引入菌株水平的差异,并使细菌丰度和代谢物之间的相关性复杂化。 SVs的复杂影响延伸到细菌和宿主表型之间的相关性。
上述两种粪便代谢物(F1P和新海藻糖)与横断面队列中个体的空腹血糖水平呈显著负相关(图3f,g)。 F. saccharivorans 的丰度与血糖显著相关(Spearman ρ=-0.38, p=1e-4, FDR=2e-4),并且在个体中,位点处存在SV(注释为K03655,假定编码ATP依赖性DNA解旋酶RecG)定义了与血糖无显著相关性的 F. saccharivorans 亚种/菌株(图3h); A. rectalis 中K01193处的SVs也导致细菌丰度和葡萄糖之间的关联系数降低,尽管在SV0组中这种关联较弱(p=0.05),这可能是亚组样本量较小的结果(图3i)。据报道,F1P可以竞争性抑制肝脏磷酸化酶,后者将糖原代谢为葡萄糖,因此粪便中的F1P可能有助于降低血糖;然而,新海藻糖与血糖的相关性尚不清楚。因此,我们的研究结果表明,通过控制使细菌丰度和代谢物浓度之间的相关性复杂化的SVs效应,结合SVs可以提高细菌和宿主健康表型相关分析的检测能力。
5 群落水平上高度相关的原噬菌体和CRISPR结构
将噬菌体整合到细菌基因组中(形成原噬菌体)和去除现有的原噬菌体都可能引入SVs,我们通过混合组装改进的宏基因组有助于原噬菌体的鉴定。使用基于机器学习的工具ProphageHunter和9612个MAGs(去除冗余之前)作为输入,我们共识别了2247个原噬菌体,基因组大小在1236-91792 bp之间(图4a)。基于连接衣壳蛋白和末端酶大亚基的系统发育分配将原噬菌体分为两个主要的病毒科:Siphoviridae和Myoviridae。此外,我们还利用长ONT reads确认了原噬菌体元件与侧翼宿主细菌基因组之间的直接联系,并建立了噬菌体家族和细菌属之间的关联,包括1077对噬菌体-宿主关联(图4b)。其中,只有72个(6.69%)被纳入当前的微生物-噬菌体相互作用数据库MVP22。
相比之下,基于短读长的宏基因组分析只发现了1815个原噬菌体,占混合组装的80.77%,这表明ONT改进的宏基因组有助于原噬菌体的发现。 除噬菌体外,微生物基因组还包含用于防御噬菌体再次感染的CRISPR-Cas系统。现在我们了解到,这些系统的基因座具有记录某些噬菌体标记序列的间隔区,从而导致同一物种内的插入/缺失变异。在上述原噬菌体分析中使用的同一组MAG中,我们还发现了150,058个CRISPR间隔区,每个宏基因组样本平均有1665±560个(平均值±SD)间隔区,这些间隔区的平均长度为34±4.8 nt(补充图9)。
而且大多数间隔区不在目前报告的数据库中,因为在CRISPROpenDB中仅发现17,600个或11.73%的间隔区,22,962个或15.30%的间隔区与西方人群肠道微生物组中的间隔区重叠(补充图10)。在本研究中,改进的宏基因组组装再次证明了发现特定基因组元件(如CRISPR间隔区)的能力增强,而基于短读长的宏基因组组装的相同分析显示只有9542个间隔区(6.36%,低15倍),这可能是由于短读长难以解析间隔区之间高度相同的重复序列。 原噬菌体和CRISPR间隔区的广泛多样性有助于定义人体肠道微生物组信息丰富、个性化的SV指纹。
事实上,我们发现,本研究横断面队列中的个体间差异显著高于同一个体内差异(在我们的时间序列队列中),这是通过基于原噬菌体/CRISPR间隔区的β距离测量的(使用原噬菌体计算Jaccard距离:Wilcoxon检验p<2e-16;使用CRISPR间隔区:Wilcoxon检验p<2e-16)(补充图11)。进一步比较原噬菌体和CRISPR间隔区在群落水平上的组成,揭示了有趣的协变。首先,Procrustes分析研究了不同类型群落差异之间的相关性,发现它们在横断面队列中不同个体的组成之间存在显著相关性(Procrustes r=0.994, p < 0.001,图4c)。其次,从宏基因组reads中识别活性噬菌体的进一步分析显示,在2247个已识别的原噬菌体中,只有47个具有潜在活性(游离),表明大量相对无活性的原噬菌体被纳入细菌基因组,构成了稳定的SVs。
图4 ONT改进的宏基因组在人体肠道微生物组中包含高度多样化的原噬菌体和CRISPR间隔区
a从200个样本中发现了228种具有完整主衣壳蛋白(MCP)和末端酶大亚基(TLS)蛋白的原噬菌体的系统发育分布,其长度分布在2.9-86 kb之间。对于每个序列,所分配的病毒家族用颜色表示,每个原噬菌体的长度显示为外圈中的条形长度。b通过分析原噬菌体序列和侧翼区域确定的原噬菌体-宿主对。原噬菌体在科水平上分组,细菌在属水平上分组,每侧显示所有序列中科/属的百分比。c Procrustes分析横断面队列(左)和时间序列队列(右)中原噬菌体/CRISPR间隔区结构,显示其整体组成之间的显著相关性,并表明同一个体在不同时间点内具有高度稳定的原噬菌体/CRISPR间隔区结构。两种统计检验的p值均为0.001。
讨论
最近利用测序技术和分析方法发展的研究强调了结构变异在人类和其他动物、各种植物以及最近的细菌中的普遍性和功能重要性。与单核苷酸多态性(SNPs)相比,结构变异在基因组中的发生率较低,但在真核生物中有更高的机会影响基因功能。然而,微生物中SVs的分析仍然具有挑战性,特别是基于短读长和映射的SVs发现高度依赖于高质量参考基因组,并且在识别大量插入和倒置方面面临困难。
结合来自ONT的长读长提高了组装质量,并支持结构变异的reads水平验证。例如,在我们对人体肠道微生物组进行的大规模测序中,我们实现了宏基因组组装,其contigs中的N50值增加了三倍以上,而基因组分箱中的N50值几乎翻了一番,超过了仅使用短读长的100 kb。有几项研究已将ONT测序应用于微生物组研究,主要重点是改进组装,每个样本需要>200 Gb的reads,这对于群体研究尚不可行;我们的研究首次报道了一项关注结构变异以及与代谢影响的关联的队列研究。与之前主要关注缺失的人类肠道微生物组研究相比,我们的分析还发现了大量插入(几乎等于缺失的数量)和数千个倒置,这极大地扩大了可检测SVs的范围。 我们的研究结果揭示了细菌类群之间基因组结构多样性的异质性,并再次强调了个体间的高度多样性与同一个体内的高度稳定性,进一步支持了SV可以作为指纹来区分不同个体肠道微生物组的观点。
在关于肠道微生物组结构变异的三项研究中,两项是横断面研究,一项研究比较了间隔三年收集的样本,结果表明SVs在同一个体中发生了显著变化。在我们的研究中,同一物种的基因组结构保持稳定,但我们承认,我们的研究无法确定一个合适的时间窗口来区分短期(10天)和长期(3年)以观察结构变异的发生。就潜在功能而言,我们研究中的SVs影响了代谢途径中富集的基因的完整性,以实现不同的养分利用、运输系统和环境感知,从而可能影响或多样化代谢能力,从而使其能够在同一细菌物种中占据和竞争生态位。结合我们研究中收集到的代谢组数据,我们进一步确定,SVs除了作为代谢活动和代谢组基础的细菌丰度外,还作为肠道微生物组变化的另一层,影响了我们筛查中约15%的粪便和尿液代谢物。
因此,SVs很可能通过调节细菌与代谢物(如新海藻糖和F1P)之间的关联,混淆了细菌与代谢物以及最终与重要宿主表型(如血糖)的相关性,从而增加了肠道微生物组与宿主健康之间关联的复杂性。然而,有必要进行进一步的功能实验来确定这些发现,因为我们的分析仍然仅限于相关性推断。我们的结果增加了观察到的微生物组SVs对代谢物和宿主表型的影响,与以色列人群和荷兰LifeLinesDEEP队列的研究结果一致,所有这些都表明考虑SVs在将微生物组与粪便/血清相关代谢组联系起来方面的效用和重要性,并最终确定宿主健康指标。 在SVs中,原噬菌体和高度可变的CRISPR元件占很大比例,我们的混合组装策略相较于仅采用短读长方法显著提高了原噬菌体和CRISPR间隔区的多样性。原噬菌体的存在为噬菌体的宿主范围和特异性提供了重要信息,而CRISPR间隔区记录了先前与噬菌体的相互作用,肠道微生物组中的新型CRISPR-Cas系统可以为未来的新基因编辑系统提供基础。我们通过识别>1000对新的噬菌体-宿主相关性来扩展当前对噬菌体-细菌宿主对的认识。
同时,在我们的数据中还发现了近6倍多的新CRISPR间隔区,这表明CRISPR间隔区的多样性仍未得到充分重视,而且研究充分的西方人群和研究较少的亚洲人群的人体肠道微生物组之间存在很大的差异。 综上所述,我们结合ONT reads的群体规模微生物组分析同时分析了人体肠道微生物组的多类型、大型结构变异。SVs调节影响宿主代谢组和健康的细菌功能,需要对细菌对人类健康和疾病的贡献进行更精细的研究,而不仅仅是关注细菌丰度。将ONT reads进一步纳入肠道微生物组研究将有助于深入解析特定时间的肠道微生物组功能,并加深我们对人类各种肠道-疾病轴的理解。
不感兴趣
看过了
取消

人点赞

人收藏

打赏
不感兴趣
看过了
取消
©2012-2023 北京华媒康讯信息技术股份有限公司 All Rights Reserved. 注册地址:北京 联系电话:010-82736610
广播电视节目制作经营许可证 —(京)字第 17437号 京海食药监械经营备20200522号
京ICP备12011723号 京ICP证150092号
 京公网安备 11010802020745号
工商备案公示信息
互联网药品信息服务资格证书((京)-非经营性-2020-0015)
京公网安备 11010802020745号
工商备案公示信息
互联网药品信息服务资格证书((京)-非经营性-2020-0015)






您已认证成功,可享专属会员优惠,买1年送3个月!
开通会员,资料、课程、直播、报告等海量内容免费看!

打赏金额
认可我就打赏我~
1元 5元 10元 20元 50元 其它
打赏作者
认可我就打赏我~
扫描二维码
立即打赏给Ta吧!
温馨提示:仅支持微信支付!
已收到您的咨询诉求 我们会尽快联系您






































 010-82736610
010-82736610
 股票代码: 872612
股票代码: 872612




