
“肾钙质沉着症”是指草酸钙(CaOx)或磷酸钙(CaPi)在肾脏中的普遍沉积。
什么是肾钙质沉着症?
What is nephrocalcinosis?
Linda Shavit, Philippe Jaeger, Robert J. Unwin,
What is nephrocalcinosis?,
Kidney International,
Volume 88, Issue 1,
2015,
Pages 35-43,
ISSN 0085-2538,
https://doi.org/10.1038/ki.2015.76.
(https://www.sciencedirect.com/science/article/pii/S2157171615321511)
Abstract: The available publications on nephrocalcinosis are wide-ranging and have documented multiple causes and associations of macroscopic or radiological nephrocalcinosis, most often located in the renal medulla, with various metabolic and genetic disorders; in fact, so many and various are these that it is difficult to define a common underlying mechanism. We have reviewed nephrocalcinosis in relation to its definition, genetic associations, animal models, and putative mechanisms. We have concluded, and hypothesized, that nephrocalcinosis is primarily a renal interstitial process, resembling metastatic calcification, and that it may have some features in common with, and pathogenic links to, vascular calcification.
Keywords: bone; calcium; hypercalciuria; hyperoxaluria; mineral metabolism; urology
关于肾钙质沉着症范围广泛,并已记录了宏观或放射学肾钙质沉着症(最常见于肾髓质)与各种代谢和遗传性疾病的多种病因和关联;事实上,这些机制如此之多,种类繁多,以至于很难定义一个共同的潜在机制。我们已经回顾了肾钙质沉着症的定义,遗传关联,动物模型和推定机制。我们已经得出结论并假设,肾钙质沉着症主要是一种肾间质过程,类似于转移性钙化,并且它可能与血管钙化有一些共同的特征和致病性联系。
严格地说,术语“肾钙质沉着症”是指草酸钙(CaOx)或磷酸钙(CaPi)在肾脏中的普遍沉积。然而,在大多数情况下,沉积似乎是间质性的,这就是肾钙质沉着症现在通常被理解的意思。虽然一些权威人士可能将肾钙质沉着症的定义限制在主要是间质性CaPi晶体的沉积上,但这并未得到普遍承认,因此,为了本综述的目的,我们保留了肾钙质沉着症的更广泛定义,以包括CaPi和CaOx沉积。当高分辨率傅里叶变换红外显微光谱和电子衍射用于研究 Randall 斑块(肾中含有间质 磷灰石 沉积物的区域,可作为尿路上皮表面 CaOx 沉积的 nidus )的组成时, CaPi 晶体被检测为其主要成分。 1然而,CaOx结石覆盖并粘附在高达50%的结石形成器中Randall的斑块上。2因此,羟基磷灰石和CaOx晶体的并发沉积可能发生在同一例肾钙质沉着症患者中。CaPi以结晶磷灰石的形式存在,但不知道CaPi的无定形沉积物是否也发生。
各种遗传性和获得性疾病与肾钙质沉着症有关,并被认为是潜在原因。肾钙质沉着症可以通过三种方式进行分类,这些方式表示肾脏受累严重程度的增加:1 分子或化学,通常在明显的高钙血症患者中观察到,并且在纠正高钙血症时通常是可逆的;2 显微镜检查,在大多数情况下是宏观肾钙质沉着症的前兆,通过在肾组织的光学显微镜下鉴定矿床来诊断;3和宏观,其中钙化在腹部 X 线平片或超声扫描上可见和/或通过计算机断层扫描确认,是肾钙质沉着症的众所周知的临床和诊断特征。虽然这些形式的肾钙质沉着症的临床表现和结果可能不同,但在临床实践中通常存在一些重叠。肾钙质沉着症通常累及肾髓质(在 97% 的患者中),或较少见的皮质。皮质肾钙质沉着症已在肾皮质坏死(通常和最初描述的产后出血后)、慢性肾小球肾炎或肾盂肾炎、原发性和继发性草症(更常见于髓质肾钙质沉着症的病因)、常染色体隐性遗传性多囊性疾病、慢性肾同种异体排斥反应和良性结节性肾钙质沉着症的患者中得到描述。本综述试图总结大量关于宏观髓质肾钙质沉着症多种病因的文献,以及它们在促进临床上显著的肾损伤中的意义。我们回顾了最近研究一些新的致病机制和进行性肾钙化的遗传背景的研究。关于明显高钙血症引起的分子性肾钙质沉着症及其后果的综述超出了本评价的范围。
肾钙质沉着症的病因
已经描述了几种新型遗传性疾病与代谢异常相关的遗传异常,这些代谢异常易导致肾钙质沉着症的发展和进展。钙转运中的上皮细胞和细胞旁紊乱导致高钙尿似乎是最重要的,同时磷酸盐或草酸盐的增加以及尿柠檬酸盐排泄的减少。此外,在某些情况下,特定的解剖异常易导致肾钙质沉着症的发展,例如,在髓质海绵肾(MSK)中。
遗传学的最新进展有助于鉴定许多参与调节肾小管钙和磷酸盐重吸收的转运蛋白、通道和受体(表 1)。参与罕见单基因疾病的几个基因与高钙质肾结石伴肾钙质沉着症有关(即CLCN5,CASR,CLDN16,CLDN19,ADCY10,SLC34A1,SLC9A3R1,GLUT2,HSPG2和FN1),而脲基调蛋白和胎儿蛋白似乎具有保护作用。.例如,在家族性良性低钙血症、新生儿重度原发性甲状旁腺功能亢进和家族性孤立性甲状旁腺功能亢进的高钙血症性疾病中,已有钙敏感受体 (CaSR) 功能丧失的突变报道,而功能获得性 CaSR 突变导致常染色体显性低钙血症伴高钙尿和V型巴特综合征。.然而,当不伴有高钙尿症时,中度高钙血症本身似乎不足以引发肾钙素沉着症,例如,在家族性良性低钙尿症患者中;只有那些有显著高钙尿症的患者才有发生肾钙沉积的风险。
表 1.髓样肾钙质沉着症(NC)相关遗传性疾病的临床表现和遗传基础
dRTA 常染色体隐性遗传 | ATP6N1B/7q33-q34,H 泵的常染色体隐性β亚基 ATP6N1B+ | 影响集合管的α插层细胞的质子分泌缺陷。低钾血症性高氯血症性酸中毒伴肾钙质沉着症和肾结石。 |
dRTA 常染色体显性遗传 | SLC4A1/17q21-q22,常染色体显性阴离子交换剂 (AE1) | 收集管的α插层细胞的基底外侧膜处碳酸氢盐运输缺陷。低钾血症性高氯血症性酸中毒、肾钙质沉着症和肾结石。 |
dRTA 伴有出生时或晚发性神经性耳聋 | ATP6B1/2cen-q13,H 泵的常染色体隐性亚基 ATP6B1+ | 集合管的α插层细胞中质子分泌缺陷。低钾血症性高氯血症性酸中毒伴肾钙质沉着症和肾结石,以及神经性耳聋。 |
巴特综合征1型 | NKCC2/15q15-q21.1,常染色体隐性 NKCC2 钠-钾-氯化物转运蛋白 | Henle 袢上行肢体的钠、钾和氯化物重吸收减少,导致低钾血症、碱中毒、高钙尿症、继发性醛固酮增多症,在某些情况下还会导致肾钙质沉着症。 |
巴特综合征2型 | KCNJ1/11q24,常染色体隐性 ROMK1 钾离子通道 | Henle 袢上行肢体的钠、钾和氯化物重吸收减少,导致低钾血症、碱中毒、高钙尿症、继发性醛固酮增多症,在某些情况下还会导致肾钙质沉着症。 |
常染色体显性遗传性甲状旁腺功能减退症 Bartter 综合征 5 型 | CASR/3q13.3-q21,常染色体显性遗传性钙敏感受体(激活突变) | 抑制 Henle 袢上行肢体的钙重吸收,导致高钙尿症、低钙血症、高磷血症和低磷食症,在某些情况下还会导致低钾血症。在某些情况下,肾钙质沉着症。 |
家族性低镁血症伴高钙尿症和肾钙质沉着症 (FHHNC) | CLDN16/3q27,常染色体显性遗传克劳丁16 | 镁和钙的尿丢失伴肾钙质沉着症,纯合子进行性肾衰竭;杂合子可能只产生肾结石。 |
家族性低镁血症伴高钙尿症和肾钙质沉着症伴眼部损伤 | CLDN19/1p34.2,常染色体显性遗传 claudin 19 | 镁和钙的肾衰竭,肾钙质沉着症和纯合子的进行性肾衰竭;黄斑疣、近视和眼球震颤。 |
常染色体显性遗传性低钙血症伴高钙尿症 (ADHH) | CASR 激活突变,常染色体显性遗传 | 低钙血症、高钙尿症、PTH 正常、复发性肾结石和肾钙质沉着症,特别是在补充维生素 D 和钙治疗期间。儿童在压力期间可能出现癫痫发作和神经肌肉烦躁;在某些情况下血清镁含量低。 |
遗传性低磷血症性佝偻病伴高钙尿症 (HHRH) | SLC34A3/ 2c (NPT2c),常染色体隐性钠依赖性磷酸盐共转运蛋白 | 肾脏磷酸盐消耗、低磷血症性佝偻病、腿部弯曲、身材矮小、1,25(OH)2D 水平升高、高钙尿症、肾钙质沉着症和肾结石。 |
登特氏病(也称为登特-1) | CLCN5/Xp11.22,内体膜上的X连锁隐性氯化物通道5 | 近端肾小管中的多个重吸收缺陷与肾滴痛、肾钙质沉着症以及许多病例终末期肾病 (ESRD) 相关。 |
洛氏综合征(也称为Dent-2) | OCRL1/Xq26.1,X-连锁隐性磷脂酰肌醇 4,5-二磷酸 5-磷酸酶 | 近端肾小管(参见 Dent's)肾成岩、肾钙质沉着症和许多终末期肾功能衰竭的多发性重吸收缺损。眼积水,白内障,智力迟钝(在Dent-2变异型中较少见)。 |
X连锁低磷血症 (XLH) | PHEX,X-连锁磷酸盐调节内肽酶 | XLH、ADHR 和 ARHR 的治疗包括长期口服磷酸盐和骨化三醇,可因间歇性高钙血症和高钙尿症而并发肾钙素沉着症(高达 80% 的 XLH 患者)。 |
常染色体显性遗传性低磷血症性佝偻病 (ADHR) | FGF23,常染色体显性遗传 | |
常染色体隐性隐性低磷血症性佝偻病 (ARHR) | 牙本质蛋白 1 (DMP1)、ENPP1 或 FAM20C | |
麦吉本-卢宾斯基综合征(ERS,牙釉质肾综合征) | FAM20A/17q24,常染色体隐性遗传 | 特征性牙齿缺陷(无色不全,牙龈增生,牙齿萌出受损)和肾钙质沉着症。 |
原发性高草酸尿症 (PH) 1 型 | AGXT/ 2q36-37, 丙氨酸乙醛酸氨基转移酶 | 50% 的成年早期患者肾钙质沉着症、肾功能损害和 ESRD。系统性草症的非肾脏表现包括心脏传导缺陷、骨痛、骨折风险增加和视力下降。 |
原发性高草酸尿症 2 型 | GRHPR/9p11,乙醛酸还原酶/羟基丙酮酸还原酶 (GRHPR) 活性降低或缺失 | PH 2 型患者的疾病通常不如 PH 1 型患者严重。患者主要表现为复发性尿石症,不太可能发生肾钙质沉着症,并且很少进展为 ESRD。 |
原发性高草酸尿症 3 型 | HOGA1,线粒体4-羟基-2-氧代戊二酸醛缩酶 | PH 3 型患者通常在生命早期(平均年龄,2 岁)出现肾结石。与其他两种形式的 PH 相反,PH 3 型患者通常在 6 岁以后没有复发性肾结石,也不会进展为 ESRD。 |
威廉姆斯-博伊伦综合征 | 7q11.23 28个基因,包括弹性蛋白基因、ELN和LIMK1 | “Elfin”面部、上脉主动脉瓣狭窄、高血压、认知受损、身材矮小、内分泌、泌尿生殖系统、听觉、牙科、眼科和皮肤科异常。发作性高钙血症和高钙尿症;>5-10%的患者肾钙质沉着症。 |
髓质海绵肾 (MSK) | 神经胶质细胞系衍生的神经营养因子(GDNF)和受体酪氨酸激酶(RET)基因散发(非遗传形式) | 复发性钙肾结石和肾钙质沉着症、高钙尿症、低枸橼尿症以及不完全和明显的 dRTA(分别为 2.9% 和高达 40% 的患者)、代谢性骨病。慢性疼痛,尿路梗阻复发和感染;然而,大多数有罕见 ESRD 报告的患者肾功能正常。 |
缩写:dRTA,远端肾小管性酸中毒;FGF23,成纤维细胞生长因子-23;PTH,甲状旁腺激素;ROMK,肾脏外髓质钾。
在Bartter综合征中已经发现了其他几个突变,通常表现为低钾血症性碱中毒,肾盐消耗,高肾碱性醛固酮增多症,高钙尿症和低citraturia(推测继发于低钾血症和钾耗竭):编码环利尿剂敏感性Na-K-2Cl(NKCC2)共转运蛋白的基因突变,肾脏外髓质钾(ROMK)通道,电压门控氯化物通道,CLC-Kb和CLC-Kb β调节亚基barttin。 肾钙质沉着症被描述为 I 型、II 型和 V 型巴特综合征的临床特征(NKCC2、ROMK 和 CaSR 突变)(表 1)。另一种与肾钙质沉着症相关的遗传性疾病是 Dent 病,其 X 连锁,其特征为低分子量蛋白尿、高钙尿症和肾结石,是 Dent 病(现称为 Dent-1),由调节近端肾小管细胞内吞作用的氯化物/质子抗转子 5 CLC-5 基因突变引起。5 此外,X连锁隐性疾病称为Lowe的眼球肾综合征,或简称Lowe综合征,这是由于OCRL基因突变引起的,4,5-二磷酸-5-磷酸酶参与肌动蛋白聚合,并且通常表现为先天性青光眼,白内障,智力迟钝以及近端小管中的多个重吸收缺陷也与肾钙质沉着症和肾结石有关;与Dent-1类似,它可能导致肾功能衰竭。还有一种称为 Dent-2 的 Lowe 综合征临床变异,主要表现为肾脏表型,与 Dent-1 相似。
家族性低镁血症伴高钙尿症和肾钙质沉着症是一种常染色体隐性肾小管疾病,通常与进行性肾衰竭和复发性尿路感染有关:它最初与claudin-16基因(也称为paracellin-1)的突变有关,claudin-16基因是claudin膜蛋白家族的成员 在各种上皮中形成细胞间紧密连接屏障,包括Henle环的厚上肢,这是这种疾病中缺陷的部位;尽管其他克劳丁突变(克劳迪恩-19)也被描述过。几种疾病已经与溶质载体家族34,成员3 SLC34A3的纯合子和复合杂合子失活突变有关,该基因编码钠(Na)依赖性磷酸盐共转运蛋白2c(NPT2c)。遗传性低磷血症性佝偻病伴高钙尿是一种常染色体隐性隐性肾磷酸盐消耗性疾病,导致低磷血症性佝偻病、腿部弯曲、身材矮小以及适当升高 1,25(OH)+2 维生素 D 水平,常伴有高钙尿、肾钙化和肾结石。19., 20., 21.最近的研究表明,具有影响两种SLC34A3等位基因的突变的个体具有显着增加的肾结石形成或髓质肾钙质沉着症的风险(46%,而在仅携带野生型SLC34A3等位基因的健康家庭成员中观察到6%)。22与一般人群相比,杂合子携带者肾钙化也更频繁,并且更可能发生在血清磷酸盐降低,磷酸盐肾小管重吸收减少和血清1,25(OH)升高的纯合子和复合杂合子个体中2维生素D水平;22然而,基因型与尿钙排泄之间没有相关性。
在成纤维细胞生长因子-23 依赖性低磷血症患者中,已发现各种遗传原因,这些疾病通常表现为显著的低磷血症,磷酸盐的肾小管重吸收阈值降低,生长迟缓,佝偻病或骨软化症,不适当正常或抑制 1,25(OH)2 维生素 D 水平、正常血清钙水平、甲状旁腺激素水平正常至高水平以及尿钙排泄正常。这些疾病包括 X 连锁低磷血症 (XLH;X 染色体上突变的磷酸盐调节内肽酶 (PHEX))、常染色体显性遗传性低磷血症性佝偻病 (ADHR;突变成纤维细胞生长因子-23)和常染色体隐性低磷血症性佝偻病(ARHR;突变牙本质基质蛋白 1 (DMP1)、ENPP1 或 FAM20C)。XLH,ADHR和ARHR的治疗是长期口服磷酸盐补充剂和骨化三醇,通常并发肾钙质沉着症(高达80%的XLH患者);这被认为是由于过度治疗活性维生素 D 或口服磷酸盐补充剂的依从性差而导致的高钙血症和高钙尿的间歇性发作的结果。最近报道的另一种遗传关联是与胰岛素受体突变有关,已知这些突变会导致严重的胰岛素抵抗,生长迟缓,脂肪和肌肉减少以及软组织过度生长,并且还与高钙尿症和肾钙质沉着症有关,但在肾脏特异性敲除(KO)小鼠模型中尚未报道。26因此,也许除了高钙尿症之外,从以肾钙质沉着症为特征的遗传性疾病中寻找一种共同的潜在或统一机制,已经提出了比提供的答案更多的问题。事实上,最近描述的 FAM20A 突变(参见上文 FAM20C)已在牙釉质肾综合征 (ERS) 中报道,这是一种罕见的常染色体隐性遗传性疾病,表现为肾钙质沉着症和特征性牙齿缺陷(不完美性牙、牙龈增生、牙齿萌出受损),其中受影响的患者是低钙性而不是高钙性。
虽然在ERS中看到的肾钙质沉着症仍然没有明确的机制,但我们提出,钙稳态的失调无论是在肾间质内局部,还是在全身上,都可能起关键作用。由于FAM20A的蛋白质产物在唾液和血液中局部分泌,并且在肾脏中也表达,因此我们认为,从头间质钙化而不是管内结晶在生物学上是合理的,可能主要是由于通常较高的钙转运和通量穿过肾小管上皮,必须防止沉积在肾间质中;因此,肾钙质沉着症可能在某种程度上类似于异常转移性组织钙化的过程。FAM20A是一种与FAM20C相关的分泌激酶,可以磷酸化酪蛋白并防止牛奶中的CaPi沉淀;FAM20C突变引起Raine综合征,其中存在异位钙化。29 FAM20C和FAM20A都可以磷酸化参与生物矿化并在肾脏中发现的兄弟姐妹蛋白MEPE(或其ASARM产物),DMP1和骨桥蛋白。30,31鉴于SHARBAND蛋白可以促进或抑制矿化,这取决于它们的形式和磷酸化状态,这种相互作用可能是牙釉质 - 肾综合征中肾钙质沉着症和牙釉质缺陷的基础。
远端肾小管性酸中毒是远端肾单位中氢离子分泌受损的一种疾病,可导致代谢性酸中毒、碱性尿 pH 值、低枸橼尿和高钙尿症伴肾钙质沉着症,以及代谢性骨病,可能是由于红细胞阴离子交换剂突变引起的 (带3,AE1)在其常染色体显性遗传形式和H-ATP酶(质子)泵的亚基突变在其常染色体隐性形式中。32., 33., 34.与 Bartter 综合征一样,远端肾小管性酸中毒中的低钾血症可能导致低枸橼尿和肾钙化倾向和结石。
另一种多系统遗传性疾病,威廉姆斯 - 博伊伦综合征,是由染色体7q11.23上1.5-1.8 Mb的半合子缺失引起的,包括∼28个基因,包括弹性蛋白基因。受累患者有多种表型,包括狭窄的面部特征、主动脉瓣上狭窄或其他血管疾病、高血压、认知受损、身材矮小以及内分泌、泌尿生殖系统、听觉、牙科、眼科和皮肤科异常。据报道,5-50%的威廉姆斯-博伊伦综合征患者经历过一次或多次高钙血症发作,这些高钙血症通常是轻度和无症状的。高钙尿症可伴随高钙血症,但也可发生孤立性高钙尿症,尤其是成人。肾钙质沉着症相对罕见,见于<5-10%的肾超声检查患者。35然而,尽管可靠地检测肾钙质沉着症的最佳影像学检查尚未确定,但现有证据表明,计算机断层扫描与超声检查或肾脏 - 输尿管 - 膀胱X射线的组合产生最准确的结果。36
在最初被认为是一种散发性疾病的MSK中,在具有发育和生长相关缺陷(如半肥大)的家庭中很少发生,在MSK中,最近的一项研究表明,50%的MSK结石患者有亲属患有较轻的MSK形式。37 在55名明显散发的MSK患者中,已经发现了主要参与肾发生发生的基因的突变或多态性,例如编码神经胶质细胞系衍生的神经营养因子和受体酪氨酸激酶的突变或多态性,支持了在“输尿管芽 - 中性间充质”处肾发生所涉及的初始事件的可能破坏 接口。虽然并非所有 MSK 患者都有神经胶质细胞系衍生的神经营养因子变异,并且有些患者没有该疾病的家族史,但可能存在遗传异质性和非遗传形式的 MSK。
最后,已经描述了几种遗传形式的原发性高草酸尿症,并与肾钙质沉着症和肾功能损害的不同发生率和严重程度相关(表 1)。最近一项针对 100 多名确诊原发性高草酸尿症患者的研究着眼于肾钙质沉着症的患病率以及结石负担与结石复发之间的关系,以及肾钙质沉着症和肾功能之间的关系。这项研究的结果表明,肾钙质沉着症的患病率为34%,其存在与更严重的肾功能损害有关。
最近在来自268个肾结石(n = 256)或孤立性肾钙质病(n = 16)家庭的272个遗传未解决个体(106名儿童和166名成人)的队列中研究了单基因疾病对肾结石疾病和肾钙质沉着症总体患病的总体患病率的贡献。在30个分析基因中的14个中共检测到50个可能的致病突变,导致所有病例的14.9%的分子诊断。成人(11.4%)和儿科队列(20.8%)的单基因病例百分比都明显较高。胱氨酸尿基因SLC7A9(n = 19)在该系列中最常发生突变。许多其他疾病已被描述与肾钙质沉着症有关。最大的临床系列是错误3在英国,由375名宏观(放射学)肾钙质病患者组成,他们在伦敦,曼彻斯特,纽卡斯尔和邓迪的40多年临床实践中收集。图 1 显示,自主性甲状旁腺功能亢进(所有患者均有原发性甲状旁腺功能亢进)、远端肾小管性酸中毒和 MSK 是该系列中最常见的相关临床诊断。
图 1.在一系列 375 例影像学 NC 患者中,肾钙质沉着症 (NC) 主要临床病因的频率。此图表基于最大的临床系列错误3 由 375 名宏观 NC 患者组成,他们在伦敦、曼彻斯特、纽卡斯尔和邓迪的临床实践中收集了 40 多年的临床经验。结果表明,自主性甲状旁腺功能亢进、远端肾小管性酸中毒(dRTA)和髓质海绵肾(MSK)是放射学NC患者中最常见的临床诊断;∼7%的患者没有明确的诊断。“其他”是指威廉姆斯-博伊伦综合征 (WBS)、巴特综合征、特发性肾 Fanconi 综合征、甲状腺功能减退、糖皮质激素抑制性醛固酮增多症和严重急性肾小管坏死 (ATN)。
肾钙质沉着症在移植后的肾脏中也有描述。反跳性甲状旁腺功能亢进和相关的高钙血症在成功肾移植后很常见,据报道,通过诱导血管收缩急性或慢性导致移植肾小管间质钙化对移植物功能产生不利影响。41 在最近一项针对 303 例肾移植受者(其中 21 例在移植时接受西那卡塞治疗)的前瞻性观察性队列研究中,肾移植中 CaPi 沉积物分别在第 3 个月和第 12 个月的移植后方案活检中观察到 33.3% 和 25.7%。42有趣的是,发现移植前甲状旁腺激素和成纤维细胞生长因子-23都是肾钙质沉着症的独立预测因子,可能与其磷酸化作用有关。然而,在本研究中,用西那卡塞特进行移植前治疗似乎既不影响移植后甲状旁腺切除术的需求,也不影响移植后肾钙质沉着症的发生率。42虽然移植后矿物质代谢紊乱可能是晶体形成倾向的原因,但暴露于潜在的肾毒性免疫抑制药物以及相关的上皮细胞损伤可能会加重晶体粘附和沉积。
肾钙质沉着症的动物模型
各种动物,包括小鼠,兔子,大鼠和猪,已被用于创建肾钙质沉着症的实验模型,试图揭示控制这种常见但复杂的疾病的机制。在大鼠中,由维生素B6消耗或给予乙醇酸,乙醛酸,草酸钠,草酸铵,乙二醇或羟基-L-脯氨酸诱导的高草酸尿症已被证明可导致CaOx在肾脏中的沉积,以及额外施用维生素D或氯化钙 加重此过程。43,44 在大多数大鼠研究中,乙二醇用于诱导高草酸尿,并导致2天内草酸盐的尿排泄增加,3天内建立高草酸尿,2周内建立CaOx结晶, 4-6周内CaOx肾结石。45 晶体首先出现在肾小管腔中,与含晶体肾小管内衬上皮细胞的损伤有关。此后,许多晶体移动到细胞间和细胞内位置,最终进入间质。46易位到间质中与炎症有关,即吸引白细胞、单核细胞和巨噬细胞,这些白细胞、单核细胞和巨噬细胞已被提议去除结晶物质。47在表面上皮脱落后,导致紧密连接松动,沉积物暴露于盆腔尿液并继续生长为大乳头状结石。
在大鼠中,高草酸尿症已被证明可增加尿液中碱性磷酸酶、γ-谷氨酰转肽酶和 N-乙酰β-葡萄糖氨酸酶的水平,可能反映近端肾小管损伤。49Tamm-Horsfall蛋白(THP)在大鼠CaOx肾钙质沉着症模型中的作用鲜为人知。在人类中,THP(uromodulin)是CaOx晶体聚集的有效抑制剂,始终存在于结石基质中;THP 的尿排泄减少已在 CaOx 或 CaPi 肾结石患者中得到证实。50然而,在具有CaOx结石的大鼠中,研究表明THP表达和产生的减少或增加。51,52
其他分子参与结晶和实验诱导的肾钙质沉着症的炎症级联反应:骨桥蛋白,各种α间抑制剂相关的大分子,比库宁,凝血酶原和硫酸肝素在肾钙质沉着症大鼠中显着增加,由其各自mRNA的表达决定。
尽管大多数具有实验诱导的高草酸尿症的大鼠会发展为CaOx肾沉积物,但肾钙质沉着症的可靠小鼠模型似乎更难产生。单独具有高草酸尿症的小鼠要么不产生或维持任何CaOx晶体沉积物,要么仅在肾脏中形成少数晶体。53,54对丙氨酸乙醛酸氨基转移酶或阴离子转运蛋白Slc26a6基因的操纵已经产生了高草酸小鼠。丙氨酸乙醛酸氨基转移酶 - 零小鼠表现出严重的高草酸尿症,但在其肾脏中仅产生少量CaOx晶体沉积物。55缺乏阴离子转运蛋白Slc26a6(近端小管中的氯化物 - 草酸盐交换剂)的小鼠也发展为严重的高草酸尿症,但只有尿钙排泄增加的小鼠才会发展CaOx晶体沉积和膀胱结石。44因此,似乎单独实验诱导高草酸尿症不足以在小鼠中产生肾钙质沉着症,并且伴随的高钙尿症和雄性性别等其他因素似乎是晶体沉积的关键因素。具有破坏磷酸钠共转运蛋白基因(Npt2a-/-) 表现出尿中磷和低磷血症的排泄增加,导致血清 1,25(OH) 升高2维生素 D 水平、肠钙通道表达增加、肠钙吸收过多、高钙血症和高钙尿症。56使用该模型,在雄性Npt2a-null小鼠中成功产生CaOx晶体沉积物。44
与CaOx结石小鼠模型不同,CaPi晶体在小鼠肾脏中的沉积似乎更容易发生;至关重要的是,这取决于THP和骨桥蛋白。THP KO 小鼠在其肾中表现出微晶形成,主要由 CaPi (磷灰石)组成,位于间隙或 基底膜区 。57,58由施用维生素D3和乙二醇诱导的高钙尿症和高草酸尿症分别导致76%的THP KO小鼠肾脏中显着的CaOx晶体沉积,而在野生型小鼠的肾脏中未检测到晶体沉积。
在一小部分骨桥蛋白 KO 小鼠中检测到肾内 CaPi 的自发性间质沉积。53骨桥蛋白和THP蛋白的缺乏导致39%的双空小鼠间质性肾结晶,与尿磷酸盐和CaPi过饱和度的升高有关。53
Npt2a基因的操作已被证明可以在不同年龄的雄性和雌性Npt2a KO小鼠中产生管状和间质CaPi晶体沉积。59此外,钠 - 氢交换剂调节因子 - 1 NHERF - 1 - null 小鼠,其 Npt2a 的表达也非常低,发展为高钙尿症,食磷过多症,高镁尿症和 尿酸尿症 ; 然而,它们在 48 - 54 周龄时在肾中仅显示少量间质 CaPi 沉积物。到72个月大时,在这些小鼠中观察到间质CaPi沉积物的显着增加。
皮质和髓质肾钙质沉着症常见于危重和低出生体重早产儿,他们接受呋塞米或维生素 D 的治疗(尽管不仅如此),以及遗传性肾小管病伴高钙尿症(如 Bartter 综合征)的儿童。呋塞米可引起肾钙质沉着症的机制归因于高钙尿症;然而,最近描述了CaPi(而不是更常见的CaOx)肾钙质沉着症的大鼠模型,其中仅氯化物或钠和氯化物消耗饮食的动物,并给予呋塞米,发展为肾钙质沉着症。在对照饮食中单独给予呋塞米没有效果,但服用不含呋塞米的氯化钠消耗饮食的大鼠也发生了明显的肾钙质沉着症。60然而,目前尚不清楚该模型中的CaPi沉积主要是皮质或髓质还是两者兼而有之。Buck等人描述的假定CaPi肾钙质病的早期大鼠模型。61 涉及定期腹膜注射葡萄糖酸钙,但钙沉积通常比髓质更皮质。因此,到目前为止,由于CaPi沉积,还没有良好的髓质肾钙质病动物模型。
总之,肾钙质沉着症动物模型的开发是一项艰巨的任务,并且这些模型不能很好地再现人类中看到的特征和关联,证实了遗传和代谢危险因素的复杂相互作用,导致钙,磷酸盐或草酸盐的尿排泄增加,以及结晶抑制剂的分子上皮转运异常,这是触发可被认为是肾钙质沉着症的过程所必需的。
肾钙质沉着症肾钙化症的机制解释
尽管遗传学研究已经为调节钙、磷酸盐和草酸盐处理的肾小管通路提供了有价值的见解,并且这些途径易导致肾钙质沉着症的代谢紊乱,例如 钙、草酸盐、磷酸盐的尿排泄增加或柠檬酸盐的排泄减少,但它们并不能完整解释导致肾钙化机制的复杂性。可以是间质性、管内或两者兼而有之。事实上,需要额外的管内因素来解释晶体潴留和对肾小管上皮的粘附:尿量减少,尿液过饱和度,存在浓度不足的晶体抑制剂,如柠檬酸盐,镁和各种蛋白质(如THP,骨桥蛋白,比库宁,尿凝血酶原) 片段1)和肾小管上皮细胞的完整性。许多实验和临床观察强烈表明,晶体不会粘附在正常分化的肾小管上皮上皮上,而是附着在表达多个晶体结合分子的去分化/再生上皮细胞上,例如含唾液酸的蛋白质和/或磷脂,磷脂酰丝氨酸,核素相关蛋白质,膜联蛋白II,骨桥蛋白和透明质酸 在肾小管上皮的腔内表面。62., 63., 64., 65. 异常上皮组织晶体粘附的因果作用已在体外肾细胞系、动物模型、肾移植患者以及骨桥蛋白和透明质酸表达与肾钙质沉着症密切相关的新生儿中得到证实。66., 67., 68.虽然这些因素可能是管内晶体形成和保留的原因,但导致间质晶体形成和沉积的特定致病机制,包括Randall斑块的发展,仍然不清楚。1然而,已经提出管内晶体易位和/或从头间质钙化。
已经提出了许多因素来解释管内晶体的形成和保留,但是导致间质晶体形成的特定致病机制(这肯定是患者肾钙质沉着症的基础)一直难以捉摸,尽管存在许多临床和遗传关联,并且它们仍然是持续争论的主题。
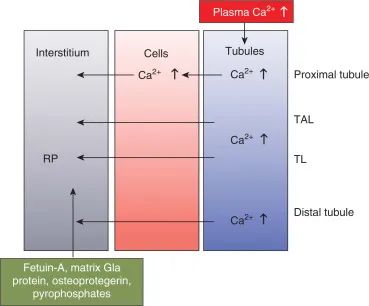
肾间质内局部钙稳态失调的失调,也可能是全身性钙质平衡失调,可能在肾钙质沉着症的发病机制中起关键作用。事实上,研究一种罕见的遗传性疾病,前面提到的牙釉质 - 肾脏综合征,表现为异常的牙釉质形成,牙齿皮屑受损和肾钙质沉着症,最近已经描绘了其中一些过程。致病基因FAM20A与钙稳态的这种全身性疾病有关,其蛋白产物FAM20A在唾液和血液中局部分泌的丰度较低。这种关联提出了这样一种可能性,即在肾钙质沉着症中,从头开始间质钙化而不是管内结晶在生物学上是合理的,并且它可能主要是由于肾小管上皮的钙转运和通量增加(图2),通常被阻止沉积在间质中,因此可能在某种程度上类似于异常转移组织钙化的过程作为起始 事件。事实上,最近一项关于小鼠肾钙质沉着症的研究表明,已知与异位矿化相关的基因的重要性,69FAM20家族分泌的蛋白激酶具有作为底物的磷脂样肽MEPE和DMP1,可以抑制骨骼和牙齿矿化,70,71并且在肾脏中也检测到72与 FAM20A 一起28 (见前文)。因此,我们认为钙化抑制剂和启动子之间的复杂失调,与血管钙化过程相似,可能在肾钙质沉着症的发展中起关键作用。在这种情况下,目前认为胎儿蛋白-A,基质Gla蛋白,骨蛋白和焦磷酸盐都以局部或全身方式起作用,以防止脉管系统钙化,31肾钙质沉着症可能代表另一种不必要的钙化过程,其中相同或相似的钙化抑制剂可以在肾间质局部发挥作用。
图 2.肾钙质沉着症间质钙化机制的简化方案。RP,兰德尔的牌匾;TAL,Henle的环的厚厚的上升肢体;TL,亨利环的细(下降和上升)肢体。
在过去十年中,许多大规模的流行病学研究为肾结石与代谢综合征,高血压,慢性肾脏疾病和心血管疾病等全身性疾病之间存在关联提供了证据。我们自己最近的研究发现,与健康的成年人相比,钙结石形成剂的腹主动脉钙化程度明显更高,72提示血管钙化可能是解释肾结石与心血管疾病之间关联的潜在机制。然而,迄今为止还没有研究解决肾钙质沉着症患者的这一问题,尽管肾间质钙化通常与肾外器官和系统疾病有关,可能指出了钙平衡异常的常见潜在过程。
不感兴趣
看过了
取消

人点赞

人收藏

打赏
不感兴趣
看过了
取消
©2012-2023 北京华媒康讯信息技术股份有限公司 All Rights Reserved. 注册地址:北京 联系电话:010-82736610
广播电视节目制作经营许可证 —(京)字第 17437号 京海食药监械经营备20200522号
京ICP备12011723号 京ICP证150092号
 京公网安备 11010802020745号
工商备案公示信息
互联网药品信息服务资格证书((京)-非经营性-2020-0015)
京公网安备 11010802020745号
工商备案公示信息
互联网药品信息服务资格证书((京)-非经营性-2020-0015)






您已认证成功,可享专属会员优惠,买1年送3个月!
开通会员,资料、课程、直播、报告等海量内容免费看!

打赏金额
认可我就打赏我~
1元 5元 10元 20元 50元 其它
打赏作者
认可我就打赏我~
扫描二维码
立即打赏给Ta吧!
温馨提示:仅支持微信支付!
已收到您的咨询诉求 我们会尽快联系您

































 010-82736610
010-82736610
 股票代码: 872612
股票代码: 872612




