
NLRP3炎症小体的激活可能会进一步提高自噬水平,从而导致炎症连锁反应。
•2022 Jan 27;13:818625.
doi: 10.3389/fimmu.2022.818625. eCollection 2022.
Role of ROS-Induced NLRP3 Inflammasome Activation in theFormation of Calcium Oxalate Nephrolithiasis
(内部资料,仅供内部学习)
草酸钙肾结石是泌尿外科常见且高复发的疾病;然而,其确切的发病机制仍不清楚。最近的研究表明,由于细胞晶体反应导致的肾脏炎症损伤在草酸钙肾结石的发展中起着至关重要的作用。越来越多的研究证实,细胞-晶体反应介导的炎症可导致肾细胞炎症损伤,促进细胞内NADPH氧化酶表达,诱导大量活性氧产生,激活NLRP3炎性体,释放大量炎症因子,引发炎症级联反应,促进钙盐晶体的聚集、成核和生长过程,最终导致肾内晶体甚至结石的发展。
关键词:活性氧,炎性体,巨噬细胞,纳米颗粒,内质网应激,自噬
泌尿外科最常遇到的疾病之一是肾结石,它与环境、遗传、代谢异常等多种因素密切相关;然而,其确切发病机制仍不清楚( 1 )。过去几年,肾结石的发病率逐年上升,美国发病率达到8.8%,亚洲发病率为3.5-7.4% ( 2、3 )。此外,肾结石在药物治疗或手术治疗后的 5~10 年内复发率高达 50%,20 年内的复发率甚至高达 75%(4)。由于高发病率和高复发率,肾结石病不仅极大地影响着人类的生命和健康,而且显着增加了医疗保健系统的财政负担( 5 )。因此,探索肾结石的确切发病机制,寻找新的治疗靶点,在结石的治疗和预防中具有重要的理论意义和应用价值。 众所周知,肾结石的形成涉及一系列复杂的过程,包括尿液过饱和、钙盐晶体粘附、积累、成核、生长和在肾脏中的滞留( 6 )。肾脏中常见的结晶主要有草酸盐(Ox)、草酸钙(CaOx)、磷酸钙和尿酸盐,其中以CaOx结晶最为常见( 7 )。肾脏内晶体的保留和随后的生长与肾结石的形成密不可分。大量研究表明,细胞晶体诱导的炎症反应在 CaOx 肾结石的形成中起着至关重要的作用 ( 8 – 10 )。在此过程中,活性氧 (ROS) 的大量产生和 NLRP3 炎性小体的激活促进了炎症因子的成熟和释放,导致肾脏和肾小管上皮细胞 (RTEC) 发生炎症 ( 11 , 12 )。在这篇综述中,我们将详细描述由 ROS 诱导的 NLRP3 炎性体刺激引起的炎症在 CaOx 肾结石形成中的作用。 去:
ROS是指生物体在有氧代谢过程中产生的一系列活性氧簇及其代谢产物,包括超氧阴离子(O 2-)、过氧化氢(H 2 O 2)、羟基自由基(OH-)等。, 主要在烟酰胺腺嘌呤二核苷酸磷酸 (NADPH) 氧化酶 ( 13 , 14 ) 的催化下在线粒体的电子呼吸链上产生。由于其高生物活性,ROS 在许多疾病的病理生理过程中发挥着至关重要的作用 ( 15 – 17 )。许多研究表明,CaOx 肾结石的起源与 RTEC 中由氧化应激介导的 ROS 过度产生密切相关 ( 18 – 20 )。临床研究证实,丙二醛(MDA)、硫代巴比妥酸反应物质(TBARS)、α-谷胱甘肽-s-转移酶(α-GST)、β-半乳糖苷酶(GAL)和N-乙酰-β-D- CaOx肾结石患者尿液中的氨基葡萄糖苷酶(NAG)显着高于健康患者,提示肾结石的发生与ROS诱导的RTEC损伤有关( 20 )。在我们之前的研究中,我们发现在大鼠 CaOx 肾结石模型中添加牛磺酸、N-乙酰半胱氨酸 (NAC)、过氧化氢酶或超氧化物歧化酶 (SOD) 时,NADPH 氧化酶的活性以及 ROS 和 LDH 的产生减少, Ox 和/或 CaOx 晶体引发的肾组织和细胞损伤和炎症反应得到缓解,肾内晶体沉积明显减少,CaOx 肾结石形成率降低( 21 )。在进一步的实验研究中,我们发现草酸或 CaOx 晶体可以促进 RTECs 中的氧化应激。大量 ROS 的产生启动 NF-κB 信号通路,促进一系列细胞因子的分泌,诱发炎症,导致 RTEC 变性、损伤、剥落、基底膜暴露,为保护创造了有利条件。肾小管腔中的晶体,最终有利于 CaOx 肾结石的发展 ( 22 , 23 )。 炎症小体是一种多蛋白复合物,在细胞识别危险相关分子模式 (DAMP)(例如尿酸单钠和二氧化硅)以及病原体相关分子模式 (PAMP)(例如病毒和细菌)后被触发和激活 ( 24 )。炎性体家族的主要成员是 NOD 样受体 (NLR)、AIM2 样受体和 pyrin 炎性体。其中,NLRP3 炎症小体是研究最多的多蛋白复合物,主要由 NLRP3、caspase-1 和凋亡相关斑点样蛋白 (ASC) 组成 ( 25 , 26 ) )。内源性或外源性刺激后,激活的NLRP3可以募集ASC蛋白并激活caspase-1,进而诱导白细胞介素(IL)-1β和IL-18等炎症因子的成熟和释放,参与多种炎症反应。身体,已成为炎症机制研究的热门话题( 27 )。既往研究表明,CaOx 晶体可通过NLRP3/ASC/caspase-1 轴直接刺激肾树突状细胞分泌 IL-1β,同时破坏 RTECs 释放 ATP,间接激活 NLRP3 炎性体,进而刺激树突状细胞分泌 IL-1β,导致肾脏炎症反应和 RTECs 的炎症损伤( 28 )。研究还表明,敲除 NLRP3 后,饲喂高草酸饮食的小鼠肾结石形成率以及 RTECs 中 caspase-1 表达和 IL-1β 分泌均显着降低 ( 8 , 10 , 29 )。此外,NLRP3炎性体的诱导可以通过改变细胞与晶体的粘附来参与肾结石的形成。Khan等研究人员发现,ROS可以通过p38MAPK途径上调透明质酸(HA)、骨桥蛋白(OPN)和CD44的表达,改变RTECs对CaOx晶体的粘附,刺激CaOx的形成石头( 30 )。齐等人。( 31 ) 发现NLRP3炎性体的诱导在ROS通过p38MAPK信号通路改变细胞对晶体的粘附和促进结石形成的过程中起连接作用,磷酸化p38和c-Jun等相关蛋白发挥重要调节作用. Joshi 等人的研究。( 10 ) 表明,在接受高草酸盐饮食的 NLRP3 基因缺陷小鼠中,RTECs 表面 OPN、HA 和 CD44 的表达水平显着降低,CaOx 结石的形成速度显着降低。和正常老鼠一样。 已经确定 ROS 是激活 NLRP3 炎性体的关键成分 ( 32 , 33 )。ROS抑制剂的应用可以阻止NLRP3炎症小体的激活,减轻细胞炎症损伤,延缓疾病的进展( 34 , 35 )。体内外研究表明,CaOx 晶体能够诱导肾脏产生 ROS,进而激活 NLRP3 炎性体,对 RTECs 和肾组织造成炎症损伤,从而促进 CaOx 结石的形成( 10 )。我们在之前的研究中发现,阿托伐他汀治疗可下调 ROS 的产生,抑制 NLRP3 炎性体通路的诱导,减少 IL-1β、IL-6、IL-18 和肿瘤坏死因子-α (TNF-α) 的释放,以及改善 CaOx 晶体在大鼠肾组织和 HK-2 细胞中引起的炎症损伤和晶体沉积 ( 36 )。因此,我们认为草酸或 CaOx 晶体在 CaOx 肾结石形成过程中诱导 RTECs 产生 ROS,并介导 NLRP3 炎性小体的活化。这一过程导致炎症细胞浸润和RTEC变性坏死,并引起肾脏炎症级联效应,促进CaOx晶体的粘附、积累、成核和二次生长,最终形成肾结石。 去:
在细胞晶体炎症反应中,巨噬细胞作为具有吞噬功能的重要炎症细胞,参与肾脏中 CaOx 晶体的形成( 37 )。发现肾结石患者的肾乳头钙化斑块组织中巨噬细胞相关基因的表达显着高于没有肾结石的患者( 38 )。已在体外报道的结果研究表明,在CaOx晶体的刺激下,肾细胞产生多种炎症因子,通过胞饮作用诱导单核细胞或巨噬细胞迁移到结石晶体沉积和转运的地方。同时,该晶体能促进细胞内NADPH氧化酶的表达,产生大量的活性氧,诱导核因子κB信号转导通路,产生大量的炎症因子包括TNF-α、IL -6 , 和 IL-1β。这进一步对肾细胞造成炎症损伤,导致细胞变性、坏死、基底膜暴露等,有利于肾钙化斑块的形成( 39、40 ))。在高草酸尿大鼠模型中,高草酸尿可通过ROS激活NF-κB信号转导通路,诱导肾脏表达大量的炎症因子,包括MCP-1、IL-6、IL-1β。在这些过程之后,会发生炎症细胞浸润(如巨噬细胞)和肾间质损伤,为肾脏中 CaOx 晶体的形成创造条件 ( 41 )。这表明 ROS 介导的巨噬细胞晶体炎症反应在 CaOx 肾结石的形成和发展中起关键作用。 在静息状态下,NLRP3在树突状细胞、巨噬细胞等先天免疫细胞中的表达水平较低,但经过内源性和外源性刺激后,激活的NLRP3可以募集ASC蛋白,激活caspase-1,进而诱导成熟和释放炎症因子,包括 IL-18 和 IL-1β ( 27 )。据报道,巨噬细胞内化病原体或晶体后可导致细胞内溶酶体破裂。溶酶体破裂后,溶酶体中的蛋白酶(如组织蛋白酶 B)被释放到细胞质中( 42 ),导致效应细胞膜离子通道的开放,引起细胞内离子浓度的变化,从而促进NLRP3炎性体的活化。羟基磷灰石晶体可通过活性氧激活NLRP3受体,刺激人单核细胞巨噬细胞和大鼠巨噬细胞产生炎症因子IL-1β( 43 )。巨噬细胞吞噬CaOx晶体后激活NADPH酶产生ROS,并在肾脏中蓄积,导致OS加重;之后,ROS 可以激活炎症通路,例如 NLRP3 和 TLR4 ( 44 )。阻断 NLRP3 可以保护巨噬细胞免受草酸盐损伤( 45 )。因此,我们推测巨噬细胞吞噬CaOx晶体后,ROS的上调可能介导NLRP3炎症小体的活化,诱导炎症因子的分泌,加重肾组织和RTECs的炎症损伤,从而促进炎症小体的形成过程。氧化钙肾结石。 高迁移率族框 1 (HMGB1) 是一种重要的炎症介质和炎症细胞因子,广泛存在于各种细胞中。它可以被激活的单核巨噬细胞和受损或坏死的细胞主动或被动地释放,以触发启动、维持和增强炎症反应的生物学效应 ( 46 )。研究发现抗HMGB1抗体的应用可显着减少肾小管间质中性粒细胞和单核细胞的浸润,减轻RTECs的炎症损伤,抑制肾组织中MCP-1、TNF-α等炎症因子的释放。和 IL-6。通过阻断 HMGB1 介导的炎症级联反应,可以改善肾功能,减轻肾缺血再灌注损伤( 47 )。王等人。( 48 ) 报道,在患有含钙肾结石的患者中,他们的尿液中发现 HMGB1 和 MCP-1 的表达增加。体外研究结果表明,高钙离子刺激 RTECs 激活 HMGB1-RAGE/TLR4-NF-κB 信号通路,刺激炎症因子 TNF-α、IL-1β 和 IL-6 的分泌( 49 )。此外,以往的研究也表明,HMGB1 的分泌与 NLRP3 炎症小体的激活之间存在密切关系 ( 50 , 51 )。拉姆坎菲等人。( 46 ) 发现脂多糖诱导的活化巨噬细胞分泌 HMGB1 需要 NLRP3、ASC 和 caspase-1 炎症复合物的参与。 综上所述,我们推测巨噬细胞吞噬 CaOx 晶体后,细胞内氧化应激产生的 ROS 通过激活 NLRP3 炎性小体、诱导 HMGB1 分泌增加、激活 NF-κB 信号转导通路来刺激巨噬细胞分泌炎性因子,包括TNF-α 和 IL-1β。另一方面,HMGB1本身又可以反过来激活NLRP3炎症小体,形成炎症的正反馈作用,从而维持甚至放大炎症级联反应,对肾脏造成严重的炎症损伤。这促进了 CaOx 晶体的粘附、积累、成核和随后的生长,并最终导致在肾脏中形成晶体甚至结石。 去:
兰德尔斑块是一种位于肾乳头黏膜下的钙化组织 ( 9 )。它起源于髓袢细长部分的肾小管上皮细胞基底膜,逐渐延伸至肾髓质间质,最后沉积在肾乳头的间质组织中( 52 )。马特拉加等人。( 53 ) 采用内镜技术观察 23 例特发性 CaOx 结石患者的肾乳头斑块,发现 24 个肾脏 (24/46) 和 156 个肾乳头 (156/172) 有 Randall 斑块,表明大多数 CaOx 结石患者有 Randall他们的肾脏有斑块。在肾小管基底膜附近,兰德尔斑的主要成分是磷酸钙。随着逐渐向肾小管腔迁移,斑块成分呈现明显的“磷酸钙→磷酸钙和CaOx→CaOx”的迁移转化现象( 1 )。学者认为,远端肾小管分泌氢气可使尿液酸化,使磷酸钙离子活性显着降低,促进磷酸钙在肾小管内溶解,释放出钙、磷离子( 54 )。在收集小管末端的最终尿液中,钙离子与草酸离子结合形成 CaOx ( 55 )。 随着现代分子生物学和细胞生物学技术的发展,Kumar 等人。( 56 )发现兰德尔斑块中存在许多与钙化纳米颗粒(CNPs)非常相似的圆形或准圆形电子高密度小体。虽然这些高密度体是否是CNPs还有待证实,但有学者表明,CNPs表面薄磷灰石壳的特征与初始阶段的Randall斑块结构高度一致( 57 )。CNPs 可以在肾结石患者的 Randall 斑周围组织、结石标本和肾盂尿中培养 ( 58 )。加西亚等人。( 59 )发现通过静脉注射或直接肾内注射CNPs均可成功构建大鼠肾结石模型。此外,Ciftcioglu 等人。( 60 )通过多种方法验证了人体肾脏组织标本肾钙化斑块中CNPs的表达,认为CNPs是肾钙化斑块的主要原因之一。因此,我们可以推断出 Randall 的斑点与 CNP 之间存在密切的相关性。 CNPs 介导的炎症与肾结石的形成密切相关。我们前期研究发现,在CNPs和RTECs共培养体系中,可以观察到RTECs粘附并吞噬CNPs,导致NADPH氧化酶活性增加,刺激细胞产生大量ROS,导致RTEC炎症损伤,这反过来又会在严重的情况下导致细胞死亡( 61 )。吴等人。( 58 )报道CNPs进入细胞后可引起线粒体损伤,进而产生ROS,通过ROS-JNk信号通路介导RTECs的损伤和结石的形成。在 Shiekh 等人之后。给Wistar大鼠静脉注射CNPs,在肾脏的病理切片中观察到钙沉积,而在大鼠的肾髓质和皮质中观察到炎性细胞浸润和聚集( 62 )。聚集的 CNPs 被巨噬细胞吞噬后,可诱导线粒体损伤并产生 ROS,从而激活 caspase-1 介导 IL-1β 的分泌,从而产生炎症反应( 63 )。作为 NLRP3 炎性体的效应蛋白,caspase-1 负责将无活性的促炎细胞因子 pro-IL-1β 切割成成熟的 IL-1β ( 64 )。我们推测CNPs可能通过产生ROS激活NLRP3炎症小体,从而介导细胞-晶体炎症反应,导致肾组织炎症损伤,诱导磷酸钙-CaOx异质成核,最终导致肾内晶体的形成。 去:
内质网(ER)是广泛存在于哺乳动物细胞中的重要细胞器。它是蛋白质制造和加工的主要细胞内场所,也是维持钙稳态的主要钙库 ( 65 )。当机体受到葡萄糖缺乏、氧化应激和Ca 2+代谢紊乱的刺激时,内质网蛋白动力学失衡导致内质网应激,从而引发未折叠蛋白反应(UPR),参与病理生理过程多种疾病( 66 )。在电镜下观察时,我们团队偶然发现CaOx肾结石大鼠的肾细胞中出现了ER的一系列异常形态变化,例如增大和畸形( 67 )。进一步的研究表明,ER 应激与 CaOx 肾结石的形成有关 ( 67 , 68 ),这与杨 ( 69 ) 和其他研究团队报告的结果一致。大量证据表明,ER 应激对 CaOx 肾结石形成的调节与细胞内 ROS 的积累和 NLRP3 炎性体的激活密不可分 ( 10 , 70 )。 ROS被视为氧化应激的标志。研究报告称,ROS 在信号转导 ER 应激方面具有双重功能。在 ER 应激期间,存在于 ER 中的 NADPH 氧化酶 (NOX) 可以刺激 ROS 的产生,进而可以调节 UPR 并恢复 ER 稳态 ( 71 )。然而,如果强刺激持续或没有及时消除,则内质网压力无法缓解,内质网氧化酶 1(ERO1)将部分引发 ROS 升高( 72 )。ER 中过量的 ROS 产生会导致钙在线粒体中沉积并增加线粒体的损伤( 73 )。我们在以往的研究中发现,SOD可以缓解ROS的应激,抑制SOD会增加ROS的积累,进而加重ER应激,促进肾结石的形成( 74 )。目前已知,在存在氧化和 ER 应激的情况下刺激 ROS 对于巨噬细胞中 NLRP3 炎性体的诱导至关重要。除了 NOX4通过介导 NLRP3 激活的 ROS 激活 NF-κB 外,它还可以激活丝裂原活化蛋白激酶 (MAPK) 以刺激促炎因子的释放 ( 75 )。此外,NOX2 可以在 ER 应激期间调节 dsRNA 活化蛋白激酶 R (PKR) 的表达 ( 76 )。PKR 的自磷酸化导致NLRP3、caspase-1 和 ASC 的从头结合,这增加了炎症小体的激活。缺乏蛋白激酶受体会显着抑制 HMGB1、IL-1β 和 IL-18 的释放 ( 50 )。 此外,ER 释放的 Ca 2+可能是导致 NLRP3 炎性体激活的一般刺激物 ( 77 )。ER是储存Ca 2+的主要细胞器,Ca 2+动员在NLRP3炎性体的激活中起关键作用。过多的 Ca 2+释放导致线粒体钙超载和线粒体损伤。线粒体 ROS 的积累增加,这导致炎症小体的额外激活和 IL-1β 的产生。阻断 Ca 2+动员可以抑制 NLRP3 炎性体复合物的生成和激活 ( 78 , 79 )。C/EPB 同源蛋白 (CHOP) 是一种转录因子,被认为是 ER 应激过程中 ER Ca 2+释放的调节因子,可通过肌醇 1,4,5-三磷酸受体调节 ER Ca 2+的释放。在 ER 压力下,CHOP 的损失导致ER 释放的Ca 2+减弱,从而减少 ROS 并提高细胞存活率 ( 80 – 82 )。因此,CHOP 和 ER 应激被认为是放大 NLRP3 炎症小体活性以增加炎症反应的潜在机制 ( 83 )。 总之,我们认为ER应激可能是诱导CaOx肾结石的ROS-NLRP3信号通路的上游或中间介导机制。这种机制不受经典 UPR 通路中的 IRE1、PERK 和 ATF6 通路的直接影响 ( 84 )。然而,它可能直接影响UPR中末端信号的表达,并通过产生ROS或介导Ca 2+动员发挥作用。 去:
自噬是一种非常保守的细胞内降解途径。在饥饿、缺氧或氧化应激过程中,细胞利用溶酶体降解受损的大分子蛋白质或细胞器,从而维持细胞内环境稳态并适应微环境变化( 85、86 )。研究表明,自噬和炎症反应之间存在密切关联(87)。自噬可以通过清除炎症蛋白聚集体和下调促炎细胞因子的释放来减轻炎症反应。反之,自噬过度激活可刺激炎症小体释放大量炎症因子,加速炎症反应进程。据报道,在肾缺血再灌注损伤大鼠模型中,自噬激活可下调促炎因子 HMGB1、TNF-α 和 IL-6 的表达,增加抗炎因子 IL-10 的释放,并减少肾脏炎症损伤 ( 88 )。柯克兰等人。( 89 )揭示细胞内ROS的过度积累可直接对细胞造成一定程度的炎症损伤。此外,高浓度的 ROS 可诱导自噬过度激活,直接导致细胞死亡 ( 90 )。 ROS和炎症反应不仅可以激活自噬,而且对CaOx肾结石的形成也有重要的调节作用。我们之前的研究结果表明,CaOx 肾结石患者的肾脏自噬水平明显高于正常肾脏的患者。CaOx晶体可诱导肾小管上皮细胞产生ROS,介导自噬过度激活,而抑制自噬可有效改善CaOx晶体诱导的肾小管上皮细胞损伤,减少乙二醇引起的肾损伤和CaOx晶体沉积,从而降低速率肾结石形成 ( 90 , 91 )。段等人。( 92 ) 还发现,自噬抑制剂氯喹可通过抑制 p38 信号通路的激活和肾草酸转运蛋白 SLC26A6 的表达,减少大鼠的氧化应激损伤、线粒体损伤,并降低尿草酸排泄和肾晶体沉积,最终抑制大鼠肾脏中 CaOx 晶体的形成。孙等人。( 93 )在体外和体内进行实验表明,抗氧化剂牛磺酸的应用可以减轻由CaOx晶体引起的RTECs的氧化应激损伤。负责任的机制是通过牛磺酸上调 Akt/mTOR 信号转导通路来抑制 ROS 介导的自噬过度激活。因此,ROS介导的自噬在CaOx肾结石的形成中也起着重要作用。 自噬在与 NLRP3 炎性体激活相关的疾病中发挥双向调节作用。Saitoh ( 94 ) 等人。2008年首次报道自噬可调节炎症小体活化,敲除自噬调节基因Atg16L1后LPS可诱导巨噬细胞炎症小体活化,提示抑制自噬可刺激炎症因子IL-18的成熟和释放和 IL-1β。柯等人。( 95 ) 报道应用自噬激活剂雷帕霉素增加自噬水平可通过消除线粒体中的 ROS 抑制 NLRP3 炎症小体的活化及其介导的炎症反应。然而,其他研究表明,ROS介导的自噬过度激活可以促进NLRP3炎症小体的激活和IL-1β的产生。张( 96 ) 研究表明,机械通气刺激肺巨噬细胞产生线粒体 ROS 后可介导自噬过度激活,而机械通气导致的肺部炎症损伤是由自噬介导的 NLRP3 炎性小体的激活引起的。信号和促炎细胞因子如肺巨噬细胞中 IL-18 和 IL-1β 的分泌。邱等人的一项研究。( 97 ) 表明三氧化二砷 (As 2 O 3 ) 可诱导肝细胞过度自噬并刺激 NLRP3 炎性体的活化,而抗氧化剂牛磺酸可改善 As 2 O 3诱导的肝细胞炎症反应。通过抑制自噬-炎症小体通路。此外,诱导NLRP3炎性体也可以抑制或促进自噬。通过激活炎症小体,尤其是NLRP3炎症小体,可以抑制线粒体自噬水平,影响线粒体的自我清除,从而促进疾病的发生和发展( 98 )。Allaeys ( 99 ) 等人。据报道,尿酸钠晶体可以通过上调 NLRP3 炎性体激活来积极管理细胞中自噬体的形成。 因此,我们推测线粒体源性ROS介导的自噬过度激活可能触发NLRP3炎性体通路,上调炎性因子IL-18和IL-1β的分泌,导致炎性细胞侵袭和肾间质炎性损伤。在 CaOx 肾结石形成过程中。NLRP3炎症小体的激活可能进一步提高自噬水平,导致肾脏发生炎症连锁反应,加速肾结石的形成过程。 去:
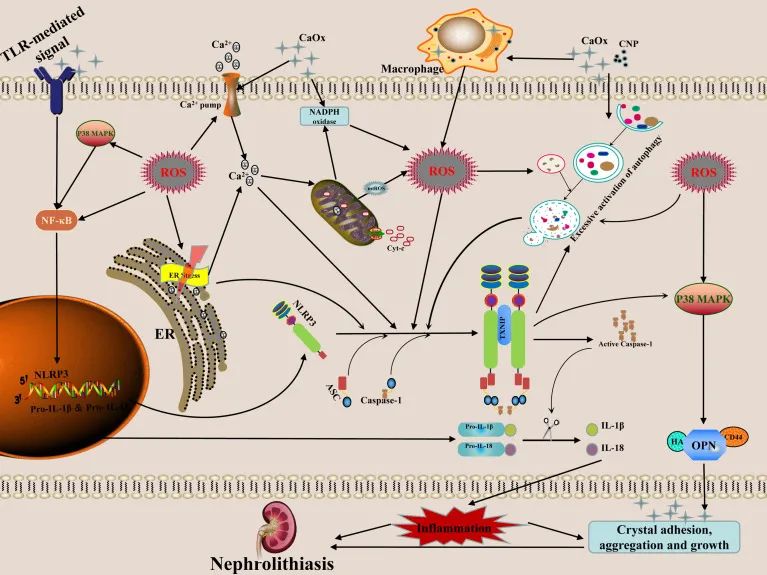
近年来,随着现代生物学技术的发展,细胞晶体炎症反应理论在CaOx肾结石形成中的作用越来越受到重视。越来越多的文献表明,RTECs 晶体反应、巨噬细胞晶体反应、钙化纳米颗粒、ER 应激、自噬激活和其他调节因子可在 CaOx 肾结石形成过程中诱导 ROS 产生。这些过程还可以介导NLRP3炎性体的激活,促进IL-1β和IL-18等炎性因子的释放,引起炎性细胞浸润和肾小管上皮细胞变性、坏死、肾炎级联效应,从而刺激肾CaOx 晶体的粘附、聚集、成核和随后的生长, 图1 )。综上所述,有效干预ROS诱导的NLRP3炎症小体激活可能是预防CaOx肾结石形成和复发的潜在治疗靶点,具有重要的理论意义和实用价值。
图1草酸钙肾结石形成中 ROS 诱导的 NLRP3 炎性体激活的可能机制和调控。成熟形式的 IL-1β 和 IL-18 的分泌是 NLRP3 炎性体激活的结果。这些介质具有促炎激活的特性,进而促进晶体的粘附、聚集和生长。晶体或纳米颗粒与细胞的相互作用会引起线粒体损伤,增加NADPH氧化酶活性,进而产生ROS,通过ROS依赖的NF-κB和自噬信号通路介导NLRP3炎症小体的转录和激活。ER通过诱导ROS产生压力期间的 NOX4 和 ERO1。ER中Ca2+的释放导致线粒体损伤,进一步加剧了ROS的释放。高浓度的ROS可诱导自噬过度激活,从而刺激炎性体释放大量炎性因子。NLRP3炎症小体的激活可能会进一步提高自噬水平,从而导致炎症连锁反应。
不感兴趣
看过了
取消

人点赞

人收藏

打赏
不感兴趣
看过了
取消
©2012-2023 北京华媒康讯信息技术股份有限公司 All Rights Reserved. 注册地址:北京 联系电话:010-82736610
广播电视节目制作经营许可证 —(京)字第 17437号 京海食药监械经营备20200522号
京ICP备12011723号 京ICP证150092号
 京公网安备 11010802020745号
工商备案公示信息
互联网药品信息服务资格证书((京)-非经营性-2020-0015)
京公网安备 11010802020745号
工商备案公示信息
互联网药品信息服务资格证书((京)-非经营性-2020-0015)






您已认证成功,可享专属会员优惠,买1年送3个月!
开通会员,资料、课程、直播、报告等海量内容免费看!

打赏金额
认可我就打赏我~
1元 5元 10元 20元 50元 其它
打赏作者
认可我就打赏我~
扫描二维码
立即打赏给Ta吧!
温馨提示:仅支持微信支付!
已收到您的咨询诉求 我们会尽快联系您
































 010-82736610
010-82736610
 股票代码: 872612
股票代码: 872612




