
功能失调的PC的存在与失调有关,代表IBD患者的一个共同特征,独立于遗传风险等位基因,PC功能障碍被认为是肠道炎症的起源。
生科云网址:https://www.bioincloud.tech
编译:微科盟听雪斋,编辑:微科盟茗溪、江舜尧。
微科盟原创微文,欢迎转发转载,转载须注明来源《微生态》公众号。
导读
论文ID
原名:XIAP restrains TNF-driven intestinal inflammation and dysbiosis by promoting innate immune responses of Paneth and dendritic cells
译名:XIAP通过促进潘氏细胞和树突状细胞的先天免疫反应抑制TNF驱动的肠道炎症和失调
期刊:Science Immunology
IF:17.727
发表时间:2021.11.5
通讯作者:Monica Yabal
通讯作者单位:德国慕尼黑工业大学
DOI号:10.1126/sciimmunol.abf7235
实验设计
研究结果
1 Xiap-/-小鼠的回肠炎症取决于微生物组
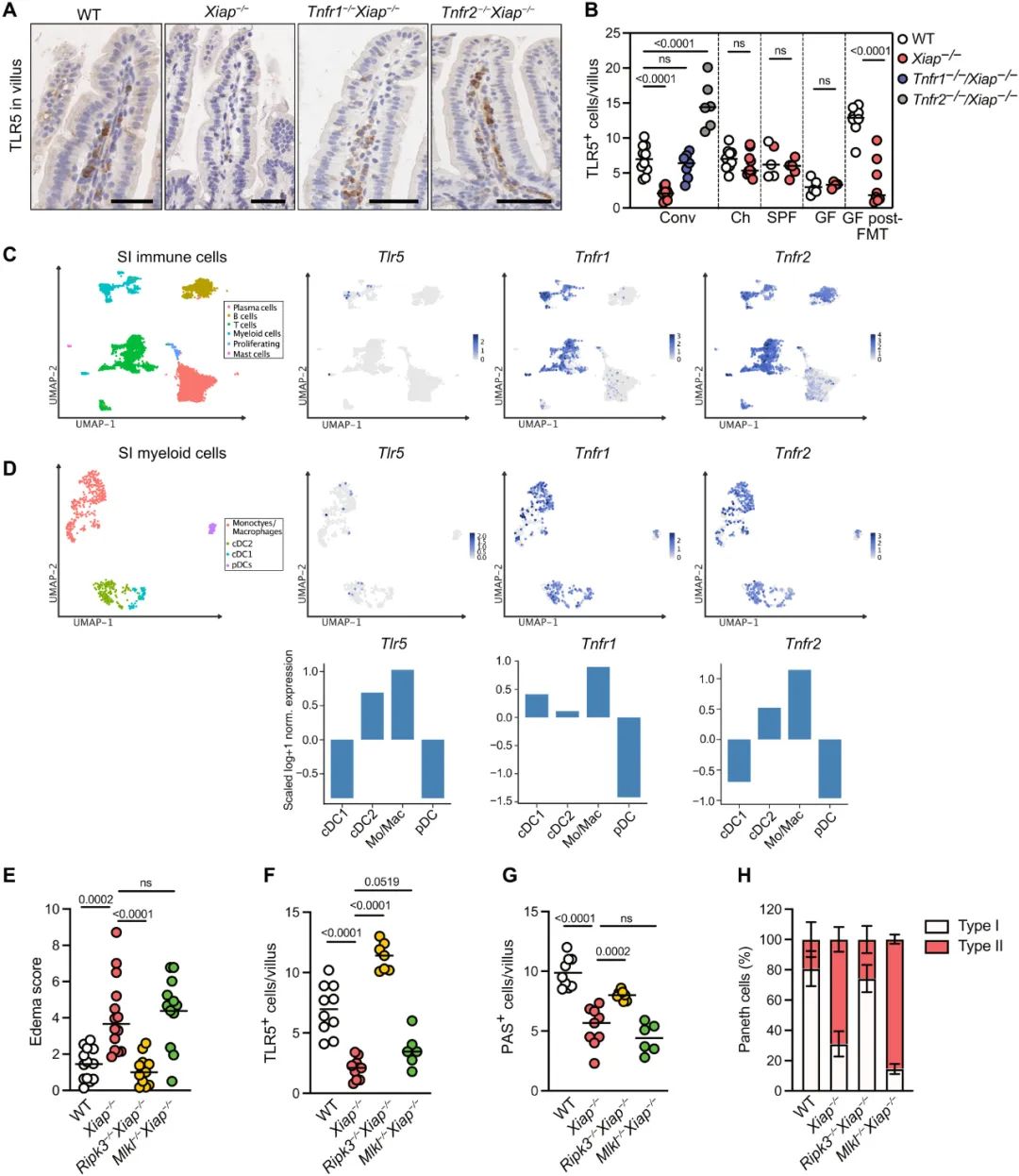
为了评估XIAP在IBD发展中的作用,我们利用了XIAP−/−小鼠模型。饲养在常规设施中的8至14周龄Xiap-/-小鼠的小肠和大肠的组织学检查显示自发性上皮下绒毛水肿(图1、A和B),特别是回肠末端,上皮增生增加(图S1、A和B)。我们还观察到这些小鼠产生的SI上皮类器官过度增殖(图S1C)。然后,我们量化了沿隐窝绒毛轴长度产生粘液的杯状细胞的数量,并观察到Xiap−/−小鼠显著减少(图1C和图S1D)。此外,相对于野生型 (WT) 动物,Xiap-/-小鼠粘膜中CD3+T细胞的浸润增加(图1D)。这些发现表明,与饲养在同一设施中的WT动物相比,Xiap-/-小鼠的炎症水平增加。
相比之下,在更严格的卫生标准下饲养的Xiap-/-小鼠(以下称为SPF小鼠)未显示任何炎症迹象(图1,A至D)。用异硫氰酸荧光素(FITC)-葡聚糖分析法对上皮屏障功能进行的功能测试表明,常规饲养的Xiap-/-小鼠的上皮通透性增加,但SPF小鼠没有(图1E)。因此,我们怀疑环境/微生物线索触发了Xiap-/-小鼠的炎症。为了验证这一假设,我们还检测了无菌WT和Xiap−/−小鼠(GF-WT/Xiap)−/−) 在使用传统设施的微生物组进行定植之前和之后[GF-post-FMT(粪便材料移植)](图S1E)。正如预期的那样,GF-Xiap−/−小鼠没有表现出任何炎症迹象,而GF-Xiap−/− FMT后出现绒毛水肿,杯状细胞减少,T细胞浸润增加(图1,A至D)。由于GF-WT小鼠暴露于微生物群中不会引起任何炎症迹象,我们得出结论,在没有微生物群的情况下,Xiap缺乏不足以引起炎症。
因此,我们通过16S测序分析了来自所有饲养设施的WT和Xiap-/-小鼠的盲肠内容物的细菌组成。与WT、SPF-WT和SPF-Xiap-/-动物相比,Xiap-/-小鼠在各组中的细菌丰富度最低,不包括GF小鼠(图 1F 和图 S1G)。这是由于相对于WT 和 SPF-Xiap-/-,Xiap-/-小鼠中的厚壁菌门(尤其是梭菌类中的梭菌群IV和XIVa的几个成员)(图1G 和图S1F)普遍减少。这是值得注意的,因为梭菌多样性和丰度的丧失是克罗恩病 (CD) 患者中常见的微生物特征。
对16S数据的进一步分析表明,来自两个饲养环境的小鼠之间微生物组的关键差异在于存在多个幽门螺杆菌属成员,这些成员仅存在于来自传统环境的小鼠中(图S1H)。映射到幽门螺杆菌属的扩增子序列变体显示出与啮齿类幽门螺杆菌的近似一致性,啮齿类幽门螺杆菌是一种仅在免疫缺陷小鼠中诱导病理学的细菌物种。因此,在Xiap-/-小鼠中,即幽门螺杆菌最有可能引发肠道炎症,正如随附的论文所证明的。
图1. Xiap−/−小鼠回肠炎症取决于微生物组(A至D)代表性图片(比例尺,200 μm)和回肠切片H&E(B)、CD3染色(C)和PAS(D)的定量。对来自常规饲养(Conv)、SPF饲养 (SPF)、GF和饲养在常规设施中后 (FMT后) 的GF的WT和Xiap-/-动物的切片进行了评估。(E) FITC-葡聚糖的血清浓度作为来自常规和SPF饲养的WT和Xiap-/-小鼠肠道通透性的量度。 (F)细菌多样性(丰富度)基于来自常规和SPF饲养的WT和Xiap-/-小鼠的16S测序。数据(A)至(F)通过单因素方差分析(P<0.0001)进行分析,图中为经Holm-Šidák校正的事后分析报告的P值。(G) WT和Xiap-/-小鼠之间差异丰富的细菌分类群的分支图。显著 (α < 0.05) 根据相应的样本组进行颜色编码。不重要的分类群用白色圆圈表示。(B)到(F)中的数据显示为点图,其中每个点代表一只老鼠。
2 恢复梭状芽孢杆菌丰度减轻Xiap−/− 老鼠的肠道炎症
在Xiap-/-小鼠中观察到的炎症的另一种解释是来自传统环境的Xiap-/-小鼠中存在病原体。尽管我们在GF小鼠中的数据表明这一解释不成立,但我们还是通过进行共饲养实验进行了功能测试(图S2A)。我们发现将WT和Xiap-/-小鼠共同饲养可降低Xiap-/-小鼠的组织病理学评分,增加杯状细胞数量,减少CD3+细胞浸润,并恢复上皮屏障完整性(图2,A至D)。我们没有检测到共饲养的WT (chWT)小鼠的炎症有任何增加,因此排除了病原体作为Xiap-/- 小鼠炎症的原因。此外,16S测序证实,共饲养导致WT、chWT和 chXiap-/-小鼠的微生物组均一,细菌多样性没有显著差异(图 2E)。
与Xiap−/−小鼠相比, chXiap−/−小鼠获得了细菌多样性,主要是受梭菌种类增加的驱动(图2F)。因此,我们得出结论,即使在存在幽门螺杆菌的情况下,与WT小鼠的微生物交换也能使Xiap-/-小鼠恢复上皮功能。此外,共同饲养的小鼠的分离导致Xiap-/-炎症表型的重建,表明Xiap-/-小鼠在与WT小鼠没有持续微生物交换的情况下无法抑制炎症(图 S2、A 和 B)。总之,这些数据加强了在Xiap-/-小鼠中检测到的粘膜完整性丧失与梭菌丰度降低有关的理论。
由于梭状芽孢杆菌类的成员,尤其是Roseburia species、粪杆菌和其他毛螺菌科的成员,是丁酸盐(一种有效的抗炎短链脂肪酸)的重要产生者,我们假设丁酸盐的损失将是导致梭状芽孢杆菌数量减少的直接后果。通过核磁共振 (NMR) 进行的无偏代谢物分析和粪便内容物的比色分析显示,Xiap-/-小鼠的丁酸和 β-羟基丁酸(丁酸的代谢物)均减少(图2,G和 H)。由于丁酸盐通过抑制组蛋白去乙酰化酶来调节基因表达,我们对传统饲养动物的回肠隐窝进行了转录组学分析,以描述在宿主上皮干细胞和PC上共存引起的变化。
无监督聚类清楚地将Xiap-/-与chXiap-/-、WT和chWT小鼠区分开来(图3A和图S3A)。基因本体分析揭示了几种代谢途径以及氧化和内质网 (ER)应激反应途径的严重失调(图S3B)。Xiap-/-小鼠中花生四烯酸代谢失调通过基于质谱(MS)的脂质组学分析得到证实,该分析显示已知源自先天免疫细胞的炎症脂质介质增加(图S3C)。在Xiap-/-小鼠中, 我们发现主要由潘氏细胞和杯状细胞产生的抗菌肽和抗菌药物(Muc13、REG3、Reg3b和Reg4)的基因表达减少(图3A)。这些数据证实了梭状芽孢杆菌在Xiap-/-小鼠微生物组中的抗炎功能。总之,在Xiap-/-遗传背景下的回肠末端观察到,自发性炎症取决于微生物组,并与梭状芽孢杆菌的丧失有关。
图2. 恢复梭状芽孢杆菌丰度减轻Xiap−/− 小鼠患者的肠道炎症(A至C)水肿评分(A)、杯状细胞计数(B)以及WT和Xiap的CD3+细胞定量(C)在常规设施 (Conv) 中,并在同一环境中共同饲养WT和 Xiap-/-小鼠(chWT和 chXiap-/-)(D)所有实验组的FITC葡聚糖血清浓度作为肠道通透性的测量指标。数据通过单因素方差分析(P<0.0001)进行分析,图中报告了Holm-Šidák校正后的事后分析P值。(E和F)细菌多样性(丰富度,E)和分支图(F)基于16S测序的差异丰富细菌类群分析Xiap −/− 和来自常规饲养的chXiap−/− 小鼠。丰富度的统计显著性通过单因素方差分析(P=0.0021)确定,图中报告的P值来自经Holm-Holm-Šidák校正的事后分析。在分支图中,显著改变的谱系(α<0.01)根据相应的样本组进行颜色编码。(G和H)所有实验组盲肠内容物中的丁酸盐(G)和β-羟基丁酸盐(H)浓度。数据通过单因素方差分析(分别为P<0.0001和0.0010)进行分析,图中为经Holm-Šidák校正的事后分析报告的P值。(A)至(E)、(G)和(H)中的数据被报告为点印迹,其中每个点代表一只老鼠。a.u.,任意单位。
图3.Xiap−/−上皮中的TNF依赖性毒性由TLR5信号诱导。(A)来自WT,Xiap−/−, chWT和chXiap−/− 老鼠的SI隐窝RNA序列的选择性去调控基因的热图。(B) 基于GSE117772的上皮Tlr5靶基因点图,以洋红色突出显示TNF信号基因。(C) Tlr5 GSEA,基于GSE117772,比较WT和Xiap−/−隐窝的基因表达(上图)和Xiap−/− 与chXiap−/−(底部)。(D和E)鞭毛蛋白和Tnf刺激的WT和Xiap−/−中Tnf mRNA水平的定量,未转导的SI类有机物(D)或用CRISPR-Cas9、sgTLR5和sgMyD88(E)转导。(D)和(E)中的数据报告为点图,其中每个点代表一个独立的实验。数据通过单因素方差分析(P<0.0001)进行分析,图中为经Holm-Šidák校正的事后分析报告的P值。(F) SI类器官分化形态分类的典型显微镜图像(×20放大;刻度条,100 μm)。(G至I)按报告处理的类器官的形态计量分析(鞭毛蛋白=500 ng/ml;TNF=20 ng/ml;丁酸盐=1 mM)。(G)至(I)中的数据由至少三个独立的实验生成,每个实验包括两到三个圆顶,每个圆顶有100到200个类有机物,并由不了解基因型和治疗的实验者进行分析。使用卡方检验进行统计分析,图中报告了精确的P值。
3 Xiap-/-上皮中的TNF依赖性毒性由TLR5信号传导诱导
到目前为止,我们的数据表明宿主对共生微生物的内在反应是Xiap−/−老鼠炎症的催化剂。为了确定宿主内在反应的关键成分,我们对来自WT和Xiap−/−小鼠回肠隐窝的RNA测序(RNA seq)数据进行了进一步分析,在共饲养前后,发现大量Xiap-/-小鼠中失调的基因是TLR5信号传导的目标(图3B)。因此,我们基于公开的数据集GSE117772,为基因集富集分析(GSEA)生成了TLR5基因信号。与WT和chXiap-/-相比,Xiap-/-小鼠的TLR5特征被下调(图3C)。
我们还注意到TLR5激活的主要信号通路是TNF信号通路(图3B和图S3D)。观察到这一点很有趣,因为TNF驱动IBD,TLR5是PCs优先表达的PRR,这是一种被认为是IBD“起源点”的细胞类型。因此,我们检查了WT和Xiap−/−原代上皮类器官对TLR5配体鞭毛蛋白和TNF的反应能力。对于鞭毛蛋白,Tnf的 mRNA表达在两种基因型之间相似,而当用重组TNF Xiap-/-重组刺激时,与WT有机体相比,Tnf水平更高(图3D)。此外,我们通过CRISPR-Cas9介导的sgRNA删除Tlr5或衔接蛋白Myd88(图S3E),并检测这些类器官对鞭毛蛋白的反应能力。在缺乏Tlr5或Myd88的情况下,WT和 Xiap-/-类器官未能诱导Tnf表达(图3E)。这些数据还表明,Myd88信号对于TLR5驱动的Tnf诱导是必要且充分的。我们利用Tlr5−/−老鼠进一步验证了Tlr5对Tnf表达的重要性。在这些小鼠中,与野生型小鼠相比,末端回肠中的组合型Tnf表达显著降低(图S3F)。因此,我们假设Xiap-/-小鼠中失调的TLR5信号传导的主要后果将是失调的Tnf信号传导,从而成为这些小鼠炎症的驱动因素。
接下来,我们进一步利用上述类器官系统来检测内在Tnf的产生是否会导致Xiap−/−细胞中的上皮细胞损伤。我们用鞭毛蛋白或TNF刺激了WT和Xiap-/-类器官,并进行了描绘上皮分化的形态计量学分析(图3,F至H)。针对TLR5与细菌鞭毛蛋白的结合,Xiap−/−类器官培养显示囊性类器官的频率增加,表明上皮生长和分化丧失(图3G)。这种效应在WT和Tlr5或Myd88缺陷培养物中不存在。相反,在TNF刺激的类器官中,同样缺乏Tlr5和Myd88的Xiap−/−的类器官对治疗仍然敏感,并且分化的类肠病毒频率降低(图3H)。总之,这些数据表明,在TLR5激活的反应中,内源性TNF的产生对XIAP缺陷的上皮细胞有害。
最后,为了了解小鼠共饲养时丁酸盐的增加是如何减少Xiap−/−小鼠中的炎症,我们刺激WT和Xiap−/−含TNF的类有机物(含或不含丁酸),并进行形态计量分析。在WT类器官中,与仅受TNF刺激的培养物相比,TNF和丁酸盐的组合没有引起类器官分化的任何显著变化(图3I)。在Xiap-/-类器官中,丁酸盐和TNF的添加显著减少了囊性类器官的数量。总之,这些数据表明chXiap-/-小鼠中梭菌增加减少炎症的一种可能机制是通过抑制TNF对Xiap-/-上皮细胞的负面影响。
4 两种TNFR均会导致Xiap-/-小鼠的肠道炎症和生态失调
接下来我们研究了TNF在Xiap−/−老鼠炎症中的作用。在常规设施中饲养的Tnf-/-Xiap-/-小鼠中,组织病理学分析表明,在没有TNF的情况下,组织损伤被消融(图S4,A至C)。TNF是TNFR1/2的配体,并且取决于细胞类型和环境,TNF信号转导的结果可能会有很大差异。因此,我们询问这两种受体是否在XIAP缺陷小鼠的发病机制中发挥作用。
因此,我们检查了Tnfr1-/-Xiap-/-和Tnfr2-/-Xiap-/-小鼠的炎症水平,这些小鼠也饲养在传统的小鼠设施中。出乎意料的是,任何一种受体的缺失都足以预防炎症和挽救上皮屏障的完整性(图4,A至D)。由于Xiap-/-小鼠的炎症与梭状芽孢杆菌的丧失相关,我们还对这些小鼠的盲肠内容物进行了16S测序。尽管Tnfr1缺失显著增加了细菌多样性(图4E),但Tnfr1或Tnfr2的缺乏并没有将微生物丰富度恢复到WT水平。然而,在Tnfr1-/-Xiap-/-和Tnfr2-/-Xiap-/-小鼠中,厚壁菌门和梭菌的丰度显著增加(图4,F和G)。总之,这些数据表明在Xiap−/−背景中通过任一受体消除TNF信号传导足以恢复上皮完整性并增加梭状芽孢杆菌的丰度。然而,与Tnfr2缺失相比,Xiap-/-小鼠中Tnfr1的缺失似乎对微生物组的影响更大。
图4. 两种TNF 受体都导致Xiap-/-小鼠的肠道炎症。(A 到 C)水肿评分(A)、杯状细胞计数(B)每个WT,Xiap−/−, Tnfr1−/−Xiap−/−和Tnfr2−/−Xiap−/−绒毛的 CD3+ 细胞定量(D) 所有实验组的血清FITC葡聚糖浓度。(E 到 G)基于Xiap−/−与Tnfr1−/−Xiap−/− (F) 和Tnfr2−/−Xiap−/−的16S测序的差异丰富的细菌分类群的细菌多样性(丰富度)(E)和分支图 (G) 来自传统饲养环境的小鼠。在分支图中,显著改变的谱系(α<0.01)根据相应的样本组进行颜色编码。(A)至(E)中的数据报告为点印迹,其中每个点代表一只小鼠。(A)至(E)中的数据通过单因素方差分析(P<0.0001)进行分析,图中的P值为经Holm-Šidák校正的事后分析报告的P值。
5 TNFR1 信号在Xiap-/-小鼠中诱导TLR5+ PC功能障碍
到目前为止,我们的数据表明Xiap−/−中的炎症是宿主对TLR5下游TNF驱动的微生物的内在反应的下游表现。由因为TLR5由PC表达,并且这些被认为在IBD中起中心作用,我们假设PC功能障碍在Xiap−/−老鼠中很明显。为了验证这一点,我们在FMT微生物定植之前和之后检查了GF WT和Xiap-/-小鼠的PC。通过用凝集素UEA1对回肠切片进行染色,我们观察到,与GF-WT相比,FMT定植后GF-WT小鼠的PC中UEA1+细胞的体积显著增加,而在Xiap-/- PC中未观察到任何变化(图5、A和B)。这些数据表明,Xiap-/- PC未能响应微生物触发。
在传统设施中持续维持的小鼠中,Xiap-/-小鼠的PC形态偶尔会随着囊泡含量的减少而缩小(图S5A)。因此,我们通过透射电子显微镜(TEM)进一步分析了PC的完整性,并根据超微结构形态(图5C和图S5B)确定了两种不同的PC群体:(i)I型,具有正常的线粒体形态(主要是线粒体A型)和堆叠的ER膜;(ii)II型,线粒体损伤(主要是D型线粒体)、内质网应激症状和囊泡室缺陷。与WT相比,Xiap-/-小鼠中II型PC非常频繁(图 5D)。来自WT和Xiap-/-小鼠的SI类器官的TEM在Xiap-/-培养物中也显示出扭曲的PC形态(图 S5C)。
此外,来自chXiap-/-和Tnfr1-/-Xiap-/-小鼠的PC的TEM分析显示,与Xiap-/-相比,这些小鼠中II型PC的频率降低。出乎意料的是,Tnfr2的缺失并未增加 I 型 PC(图5D)。因为我们对TNF介导的PC功能障碍的假设是基于TLR5参与的假设,所以我们观察了TLR5在回肠隐窝中的表达,特别是在PC中的表达(图5E)。半定量分析显示Xiap-/-和Tnfr2-/-Xiap-/-小鼠的PC中TLR5的表达降低,而共饲养和Tnfr1的缺失足以增加TLR5表达(图 5F)。因此,TLR5在PC中的表达减少与其细胞器形态紊乱相关。
根据这些观察结果,似乎删除Tnfr1对PC完整性的影响最大,而Tnfr2可能在其它地方发挥作用。因此,为了对这一假设进行功能测试,我们从Tnfr1−/−Xiap−/−和Tnfr2−/−Xiap−/−小鼠中生成了类器官,以评估TNF和鞭毛蛋白对其生长的影响。形态学分析表明,尽管TNF和鞭毛蛋白都抑制了Tnfr2-/-Xiap-/-培养物中的分化,但Tnfr1-/-Xiap-/-培养物仍然不受TNF和鞭毛蛋白的影响(图5G)。因此,这些数据表明Xiap-/-上皮中TLR5介导的上皮和PC功能障碍可归因于TNFR1介导的信号传导。
图5. TNFR1信号损害Xiap-/-小鼠的TLR5+ PC 功能。(A和B)回肠切片隐窝内PC中免疫荧光UEA1 染色(紫色)的代表性计算机渲染表面(A)和定量(B)GF WT和Xiap-/-小鼠在(GF)和之后(GF post-FMT)转移到常规设施之前。用 DAPI 复染细胞核(灰色)。从每个基因型和饲养条件的三只动物的一张载玻片中获得数据。通过使用Imaris软件为隐窝内的UEA1+区域生成表面来分析数据。(C) WT和Xiap-/-回肠切片中PC形态的代表性TEM图像,显示变形的囊泡、ER 膜和线粒体形态。(D) 根据WT (n = 8)、Xiap−/−(n = 8) chWT (n = 4)、chXiap−/− (n = 4) 的形态和线粒体完整性,PC分为I型和II型)、Tnfr1-/-Xiap-/-(n = 4) 和Tnfr2−/−Xiap−/− (n = 4) 小鼠来自指定的环境。数据通过单向方差分析 (P < 0.0001) 进行分析,图中报告了来自具有 Holm-Šidák 校正的事后分析的 P 值。(E 和 F)来自 WT、Xiap-/-、Tnfr1-/- 的回肠组织隐窝(n = 5至10)内TLR5信号的代表性图像(比例尺,100 μm)(E)和量化(F)Xiap-/-和Tnfr2-/-Xiap-/-小鼠来自传统环境。0分相当于没有检测到表达,3分是高表达。每只动物20个隐窝由对基因型不知情的实验者评估。(G) 来自用 TNF (20 ng/ml) 或鞭毛蛋白 (500 ng/ml) 刺激的 WT、Xiap−/−、Tnfr1−/−Xiap−/−和Tnfr2−/−Xiap−/−小鼠的类器官的形态测量分析)。数据是通过至少三个独立实验获得的,每个实验包括两到三个圆顶,每个圆顶有100到200个类器官。使用卡方检验进行统计分析,图中报告了精确的P值。
6 TNFR2信号转导TLR5+ DCs
我们将上皮和PC功能障碍归因于TNFR1信号,而Xiap−/−小鼠中TNFR2介导的炎症背后的机制仍不清楚。因此,我们寻找在小肠中表达TLR5的其他细胞类型。通过TLR5的免疫组织化学(IHC),我们在WT小鼠的绒毛和LP中检测到TLR5+细胞群,在Xiap−/−中显著减少(图6、A和B)。这些TLR5+细胞的频率因Tnfr1缺失而增加至WT水平,并因Tnfr2缺失而显著进一步增加。在共同饲养的SPF-Xiap-/-和GF-Xiap-/-小鼠中对这些TLR5+细胞的定量表明,在这些条件下,TLR5+细胞的数量与其WT对应物相当(图6B)。因此,在发炎的Xiap-/-回肠切片中,这些TLR5+细胞被耗尽。对于微生物定植,在GF-WT小鼠中TLR5+细胞的数量显著增加,但在GF-Xiap−/− 中没有增加(图6B)。这些数据表明,在小肠中,这些细胞是通过与微生物组的相互作用招募或诱导的。此外,这也意味着XIAP缺陷动物在维持这种细胞群方面存在固有问题。
以前的文献已经指出Tlr5在SI中的表达,特别是在DC中。为了明确鉴定这种细胞群,我们对从WT小鼠LP中提取的细胞进行了单细胞RNA-seq(scRNA-seq)。对来自LP的免疫细胞的初步分析表明,Tlr5仅在髓系细胞中表达(图6C)。深入分析显示Tlr5表达细胞属于2型常规DC(cDC2)和单核细胞/巨噬细胞群(图6D)。与其他髓系细胞群相比,这两个细胞群也表达了相对更多的Tnfr2(图6D)。此外,来自WT和Xiap-/-小鼠回肠组织的Tnf、Tnfr1和Tnfr2的定量聚合酶链反应 (qPCR )支持Tnfr2在促进炎症中的作用,因为与WT小鼠相比,Tnfr2的表达增加(图S6A)。总之,这些数据表明,在XIAP缺乏的情况下,TLR5参与和随后的TNF信号将主要影响cDC2和单核细胞群。
图6. TNFR2信号传导靶向TLR5+ DCs导致炎症细胞死亡。(A和B)WT、Xiap-/-、Tnfr1-/-Xiap−/−和Tnfr2−/−Xiap−/−小鼠(n = 5到10)回肠绒毛内TLR5+ 细胞的代表性图像(比例尺,100 μm)。对来自所有不同饲养条件的WT和 Xiap−/−进行了进一步分析。数据通过单向方差分析(P < 0.0001)进行分析,图中报告了来自具有Holm-Šidák校正的事后分析的 P 值。(C)从三只WT小鼠中提取的LP CD45+细胞的UMAP,显示Tlr5、Tnfr1和Tnfr2的表达。 (D) 来自 (C) 的LP骨髓细胞的指定基因的UMAP和标准化表达。(E到G) WT、Xiap-/-、Ripk3-/-Xiap-/-和Mlkl-/-Xiap−/−老鼠的水肿评分(E)、TLR5+免疫细胞(F)和杯状细胞(G)的定量。数据通过单向方差分析(对于所有比较,P < 0.0001)进行分析,图中报告的P值来自事后分析和 Šidák 校正。(H) 基于来自指定基因型的回肠隐窝的TEM对I型和II型PC进行量化,基于每个基因型每只小鼠5到10个隐窝,每个基因型4到5只小鼠。
7 Ripk3缺失可拯救Xiap−/−老鼠的回肠炎
在体外,TNFR2信号在TLR/Myd88激活的情况下诱导髓样细胞的RIPK3依赖性细胞死亡。基于此,我们假设Ripk3可能是Xiap−/−老鼠中TLR5+髓样细胞丢失的原因。我们通过评估Ripk3−/−Xiap−/−中的回肠炎症在体内证实了这一假设。在这些小鼠中,根据组织病理学评分将炎症恢复到WT水平(图6E),并且TLR5+细胞的数量显著增加(图6F)。在上皮区室中,与Xiap-/-小鼠相比,杯状细胞和I型PC的数量也有所增加(图6,G和H)。此外,我们还研究了Mlkl缺失对Xiap−/−小鼠的影响,因为Mlkl是RIPK3依赖性细胞死亡的典型效应物。然而,在 Mlkl-/-Xiap-/-小鼠中,与Xiap-/-小鼠相比,没有观察到炎症的显著改善(图6,E至H)。总之,这些数据表明RIPK3在Xiap−/−小鼠中的激活是肠道炎症的关键驱动因素,而MLKL没有明显参与。
为了模拟先天免疫细胞中TLR/Myd88参与XIAP缺陷的后果,我们利用了体外骨髓衍生 DC (BMDC) 模型,该模型由巨噬细胞和树突状细胞组成。TLR5在小鼠LP DC中的限制性表达妨碍了对BMDC中TLR5信号的研究。然而,由于Myd88激活下游TNF信号后的下游信号通路与许多其他TLR共享,该模型允许我们通过研究其他TLR触发Myd88的结果来推断TLR5激活的后果(图3、F和G)。与回肠中TLR5+细胞的模式一致,用TLR4配体脂多糖(LPS)刺激BMDC可导致Xiap−/−活性显著下降,这最好通过Tnfr2的缺失阻断(图S6D)。类似地,TLR3和TLR9的结合也诱导Xiap−/−的细胞死亡(图S6、A和B)。与体内数据一致,炎症细胞死亡依赖于Ripk3,而非Mlkl(图S6、B和C)。
为了了解Ripk3和Mlkl是否在XIAP-proficient细胞中起到类似的作用,我们通过泛半胱天冬酶抑制剂Z-VAD-fmk阻断所有半胱天冬酶,在WT BMDC中伴随TLR/Myd88信号强制诱导Ripk3。这些实验表明,在这种情况下,Tnfr2、Ripk3和Mlkl缺失最有效地防止了细胞死亡(图S6D)。因此,在 TLR/Myd88 激活的背景下,在XIAP缺陷和XIAP-proficient细胞的骨髓细胞中,依赖 RIPK3的细胞死亡需要TNFR2信号传导。有趣的是,Mlkl对Xiap−/−细胞经历细胞死亡来说是可有可无的,最有可能是由于这些细胞中已记录的caspase-8活化增加。因此,这些数据确定TLR-TNFR2-RIPK3信号轴是肠道炎症的关键引发剂,通过促进XIAP缺陷的DC和单核细胞中的炎症细胞死亡。
8 在诊断为IBD的儿童患者中,TNFR2升高与更严重的病程相关
为了评估诊断为IBD的儿童患者TNFR2表达升高的意义,我们使用了两个不同的队列,XLP2队列和RISK研究。我们生成了XLP2队列,包括7例表现为IBD(XLP2)的XLP2特征性儿童患者、8例IBD(IBD)常规儿童患者和15例非IBD健康儿童对照的粘膜活检(表S1)。非IBD对照组来自健康和无炎症的粘膜区域,这些粘膜区域是从接受阑尾炎手术的儿童患者中取样的。盲法病理检查显示,与健康对照组相比,IBD和XLP2患者组呈现出共同的病理外观(图S7、a和B)。此外,我们使用了来自RISK研究(GSE93624)的数据,其中包括27名在3年内进展为复杂疾病(cCD)的CD患者、210名治疗初期的CD儿童患者和35名非IBD对照组。
来自这两个队列的基因表达数据的基因本体论途径分析揭示了类似的炎症和代谢途径模式,这是IBD的特征(图S7、C和D)。这些途径让人想起在Xiap−/−小鼠中看到的解除管制的途径(图S3B)。通过比较cCD和XLP2之间的共性,我们确定了最重要的调控基因中的细胞因子抑瘤素M(OSM)及其受体OSMR、MMP9和CX3CL1(图7、A和B以及图S7、E和F)。这些基因被认为是复杂疾病的生物标志物,因此可以表示XLP2患者的严重病程。
为了了解TNF在疾病进展中的作用,我们进一步分析了cCD和CD队列中TNF途径的差异。我们鉴定了8个显著上调的基因,即TNF、CX3CL1、CREB3L1、IL18R1、MMP9、MLKL、TNFRSF1B和CCL5。接下来,我们构建了一个基于相关性的网络,揭示了TNFRSF1B与MMP9、TNF和MLKL之间的正相关性(图S8A)。MLKL的上调表明这些患者可能参与坏死性细胞死亡途径。对XLP2和IBD队列的类似分析确定了TNF途径中的18个上调基因以及TNFRSF1B与SELE、IL6和MMP3呈强正相关,它们是组织损伤和炎症的有力驱动因素(图S8A)。总之,这些数据表明XLP2综合征与严重炎症标志物的增加相关,而严重炎症标志物的增加与TNFR2信号的升高呈正相关。
在小鼠系统中,我们确定cDC2和单核细胞是表达TNFR2最多的髓样细胞类型(图6)。为了验证CD患者是否也存在这种情况,我们利用了基于回肠末端活检(E-MTAB-8901)的新诊断CD儿童患者(n=7)的公开scRNA序列数据集。在CD患者中,与健康对照组相比,髓样细胞室扩大/扩大(图7、C和D),我们在髓样细胞群中发现了TLR5表达细胞,而健康对照组中没有TLR5表达细胞(图7、C和D)。TLR5的高表达进一步局限于cDC2(图7E)。与小鼠系统类似,与其他髓系细胞相比,人类cDC2的TNFRSF1B表达升高(图S8B)。
总之,在XLP2和RISK患者队列中,我们观察到两个队列中TNFRSF1B表达增加与疾病严重程度标志物呈正相关。此外,验证小鼠发现,在第三个患者队列中cDC2中TNFRSF1B和TLR5的共表达表明这些患者队列中的共同分子机制是活跃的(图S8C)。在CD患者的炎症状态下,TLR5+TNFR2hi cDC2发生扩张,在XIAP缺乏时,使它们容易发生炎症细胞死亡,从而加剧小肠炎症。
图7. TNFR2表达升高与IBD中的复杂疾病相关。(A和B)TNFRSF1A、TNFRSF1B、RIPK3、MLKL、OSM和OSMR在非IBD、IBD和XLP2(A)和RISK患者队列(GSE93624)(B)回肠活检中的表达。Wald检验用于从成对比较中生成指示的P值。(C至E)根据E-MTAB-8901从健康对照组和(D)患有CD的儿童患者中分离的免疫细胞的scRNA序列,以及(E)从(D)中分离的髓样细胞。
讨论
IBD领域的一个争论点是,生物失调是疾病的原因还是结果。迄今为止,GWAS研究已经确定了易患IBD并与微生物失调发展相一致的多种遗传变异。这通常表现为微生物组内多样性减少,尤其是厚壁菌门丰度减少。目前,两个一致的概念正在出现,它们将遗传易感性与肠道微生物群中经常观察到的疾病相关变化联系起来。第一种是,生态失调本身与特定遗传功能的丧失无关,而是由于易受感染宿主的炎症反应解除管制,并且常常被夸大而导致的生态失调。第二个概念是,这些基因多态性不仅使受影响的个体易受肠道过度炎症的影响,而且还允许其他无害的环境线索和微生物群触发过度炎症。例如,这一概念基于最近的研究,在比较与CD相关的NOD2变异的患病和健康携带者的微生物组时,发现没有基因型微生物组关联;相反,生物失调与疾病状态有关。
本文提供的数据支持宿主基因调节过度炎症反应倾向的模型,而过度炎症反应最终导致微生物失调。我们在XIAP缺陷小鼠中观察到,被定义为梭状芽孢杆菌数量减少的生物失调是炎症的特征,而不是XIAP缺陷宿主的固有表型。相反,是对细菌的不适当反应导致了TNF介导的组织损伤。这些结论也得到了Strigli等人随附论文的支持。该论文研究了XIAP缺陷宿主对肝幽门螺杆菌感染的反应。这一范例得到了典型成果的进一步支持,该作品中使用了携带与成人IBD相关的两种最常见基因变体的重组小鼠,即NOD2和ATG16L。
我们关于微生物组的结论的一个警告是,我们在非共同饲养的Xiap−/−小鼠中看到了生态失调,这不能转移到chWT小鼠。因此,这些微生物变化是一种隐性特征,被共同饲养所掩盖。所以,我们得出结论,XIAP不会直接改变微生物组,但对维持共生宿主微生物组环境至关重要。
在Xiap-/-在小鼠中,组织损伤是由PC和骨髓细胞功能障碍引起的,这可以追溯到Tlr5的表达。PC是肠干细胞生态位的一部分,是高度分泌和长寿的细胞;因此,它们对内质网应激、TNF信号和自噬缺陷特别敏感,从而导致细胞死亡。此外,功能失调的PC的存在与失调有关,代表IBD患者的一个共同特征,独立于遗传风险等位基因,PC功能障碍被认为是肠道炎症的起源。然而,在没有微生物变化的非IBD患者中也观察到功能失调的PCs。这些发现支持了PC功能受损不足以发展IBD的观点。这与我们的研究结果一致,在Tnfr2缺陷小鼠中,在消除炎症和生物失调的条件下,仍能观察到功能失调的PC。
在这项研究中,尽管Tnfr1缺陷保护了PC功能和挽救了TLR5+免疫细胞数量,但Tnfr2的缺失只挽救了LP TLR5+细胞。我们的数据表明,这些TLR5+细胞是髓样细胞,最有可能是cDC2或单核细胞/巨噬细胞,它们在Xiap−/−小鼠中丢失,与已发表的Tlr5研究一致。此外,利用体外BMDC模型,我们可以证明TNFR2介导了Xiap−/−的细胞死亡TLR激活后的髓样细胞以RIPK3依赖性方式表达。因此,尽管我们无法在回肠中直接显示这一点,但我们提供的证据表明,除了调节T细胞功能外,TNFR2也在髓细胞存活中发挥作用。我们认为,这是IBD发病机制的关键,很可能是IBD与其他基因多态性相关的一个重要特征。
我们的工作强调了先天免疫系统中的多个层面的失败,这些失败最终导致TNF信号驱动的病理学。在剖析TNFRs的作用时,我们偶然发现了TLR5,这在事后看来并不令人惊讶。
在肠道中,TLR信号对共生微生物配体反应较弱,这是组织内稳态的关键机制。一个例外是TLR5,与TLR4例如相反,它在从LP分离的CD11c+细胞和PC中具有活性。因此,尽管我们的体外研究表明,多种TLR在体内诱导Xiap−/−骨髓细胞的TNF和RIPK3依赖性细胞死亡,但需要明确定义组织和细胞类型特异性TLR功能库受TNF信号影响的细胞类型。
此外,我们之前已经证明,髓样细胞上TLR的激活诱导TNFR2的上调。因此,正如我们在scRNA-seq实验中观察到的那样,推测TLR5在体内的活性与髓样细胞中的Tnfr2高表达之间存在功能联系是很有诱惑力的。TLR和TNFR2的联合活性则表现为Xiap−/−易受TNF-和RIPK3依赖性炎性细胞死亡影响的细胞。我们进一步推测,XLP2和CD患者队列中MLKL表达的对比调节反映了在缺乏XIAP的情况下其在细胞死亡诱导中的可有可无的作用,如我们在此所证明的。
在小鼠中,由于所使用的实验小鼠的基线微生物组成和卫生状况非常不同,TLR5在塑造共生生态系统中的作用仍然存在争议。例如,对Tlr5−/−小鼠的两个独立菌落的研究表明,该微生物组决定自发性结肠炎的发病。此外,Tlr5的细胞类型特异性缺失进一步剖析了其在肠道内环境稳定中的作用。肠上皮细胞中Tlr5的缺失会引起厚壁菌门和拟杆菌门比例的变化,而DC中Tlr5的缺失则不会。这与我们的数据一致,表明Tnfr1和PC在介导微生物组变化方面发挥着更为主导的作用。总之,这些数据也表明DC中TLR5的缺失不是致病性的。因此,我们假设这些细胞的炎症性死亡对宿主有害。
最后,最近的临床研究表明,对诊断为XLP2的儿童进行造血干细胞移植能够逆转肠道炎症并增加梭状芽孢杆菌的数量,从而确定免疫系统是肠道炎症的关键驱动因素。我们的模型提供了宿主对TNF的异常反应是如何引发炎症和随后的微生物失调的机制性见解。因此,这些发现可能成为未来旨在抑制肠道炎症细胞死亡的治疗干预措施的思路。
不感兴趣
看过了
取消

人点赞

人收藏

打赏
不感兴趣
看过了
取消
©2012-2023 北京华媒康讯信息技术股份有限公司 All Rights Reserved. 注册地址:北京 联系电话:010-82736610
广播电视节目制作经营许可证 —(京)字第 17437号 京海食药监械经营备20200522号
京ICP备12011723号 京ICP证150092号
 京公网安备 11010802020745号
工商备案公示信息
互联网药品信息服务资格证书((京)-非经营性-2020-0015)
京公网安备 11010802020745号
工商备案公示信息
互联网药品信息服务资格证书((京)-非经营性-2020-0015)






您已认证成功,可享专属会员优惠,买1年送3个月!
开通会员,资料、课程、直播、报告等海量内容免费看!

打赏金额
认可我就打赏我~
1元 5元 10元 20元 50元 其它
打赏作者
认可我就打赏我~
扫描二维码
立即打赏给Ta吧!
温馨提示:仅支持微信支付!
已收到您的咨询诉求 我们会尽快联系您







































 010-82736610
010-82736610
 股票代码: 872612
股票代码: 872612




