
众所周知,肿瘤具有高度的异质性,在不同癌种、不同病例、不同细胞间,都存在巨大差异。
众所周知,肿瘤具有高度的异质性,在不同癌种、不同病例、不同细胞间,都存在巨大差异。这种差异表现在基因表达水平、基因组变异水平、染色体稳定性水平等多个层面。多个层面的异质性均与差预后显著相关,同时作者也揭示特定基因和特定拷贝数变化在肿瘤发生发展中的作用(图1)。
图1:不同样本的不同变异类型的克隆占比以及其生存分析
使用组学测序来探究肿瘤的异质性被证明十分有效,例如之前大名鼎鼎的PCAWG计划,该项目从多个方面对于泛癌的特征与进展进行了细致的描述。其中,Moritz Gerstung等人通过全基因组测序分析2658个泛癌全基因组,作者重建了38种癌症的突变过程和驱动突变序列的进化过程。作者详细描述了样本间多种类型克隆分类的占比,以及克隆的进化水平。同时作者也详细发掘了不同的染色体变异以及基因SNV等在不同肿瘤发展时间段的发生情况。
图2:不同患者肿瘤进展轨迹
传统的肿瘤进化分析,基于大样本量的全基因组测序分析,然而其成本是非常昂贵的。以低廉的检测手段进行肿瘤进化分析,是研究者的关注热点。统观各类组学分析数据,只有单细胞测序转录组可以较低成本实现高通量的数据。因此,若能基于单细胞测序数据解释肿瘤的进化关系,无疑是比较经济实惠的方案设计。
单细胞可以做肿瘤克隆进化么?
对于高通量的10Xgenomics单细胞测序数据我们可以考虑使用InferCNV或者CaSpER来推断CNV的变异,使用[uphyloplot2](https://github.com/harbourlab/uphyloplot2)来进行树形推断。
首先我们选用了InferCNV对癌肿样本的上皮细胞(Marker:EPCAM)(图3)进行染色体CNV的变异推断。
图3:上皮细胞的marker表达及样本UMAP图
InferCNV,参考细胞选择的是癌旁样本的免疫细胞。可以观察到上皮细胞之间存在不同的CNV变异,由此可以根据此来推断不同恶性细胞之间的克隆及进展状况。
图4: inferCNV获得的CNV变异全景图
接下来我们uphyloplot2来进行树形的可视化工作。uphyloplot2的参数较为简单,只需要选取克隆的大小就可以。
具体的可视化结果如下:
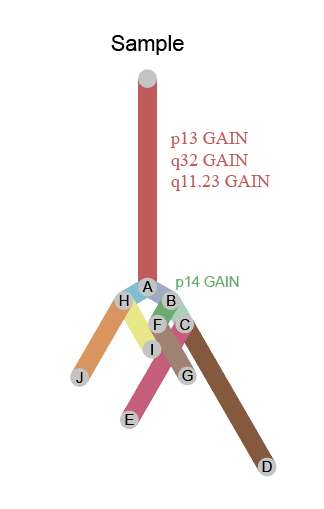
图5:克隆进化可视化(sample 为本文测试样本,test1、test2为软件自带测试数据)
当然我们有时还需要在图上加上CNV缺失的注释或者驱动基因,这部分的结果也可以在inferCNV输出的pred_cnv_regions.dat中找到,使用绘图工具加上就算初步完成癌肿样本的克隆进化的分析了。
图6:加上CNV等注释信息的克隆进化图
小结
派森诺生物单细胞产品部门在肿瘤CNV推断、克隆进化现有的多种方法上,优化总结了多套分析与可视化方案。派森诺分析团队可将肿瘤进化结果结合空间转录组数据,推出空间肿瘤进化分析。除此之外,分析团队也致力于开发方法研究多组学联合分析来探究肿瘤的进展与特征。
不感兴趣
看过了
取消

人点赞

人收藏

打赏
不感兴趣
看过了
取消
©2012-2023 北京华媒康讯信息技术股份有限公司 All Rights Reserved. 注册地址:北京 联系电话:010-82736610
广播电视节目制作经营许可证 —(京)字第 17437号 京海食药监械经营备20200522号
京ICP备12011723号 京ICP证150092号
 京公网安备 11010802020745号
工商备案公示信息
互联网药品信息服务资格证书((京)-非经营性-2020-0015)
京公网安备 11010802020745号
工商备案公示信息
互联网药品信息服务资格证书((京)-非经营性-2020-0015)






您已认证成功,可享专属会员优惠,买1年送3个月!
开通会员,资料、课程、直播、报告等海量内容免费看!

打赏金额
认可我就打赏我~
1元 5元 10元 20元 50元 其它
打赏作者
认可我就打赏我~
扫描二维码
立即打赏给Ta吧!
温馨提示:仅支持微信支付!
已收到您的咨询诉求 我们会尽快联系您





































 010-82736610
010-82736610
 股票代码: 872612
股票代码: 872612




