


















重磅研究 | Microbiome:中科院微生物所建成人肠道微生物生物库(hGMB)




本研究从239份健康中国志愿者粪便标本中分离到10558株细菌。
编译:微科盟R.A,编辑:微科盟木木夕、江舜尧。
微科盟原创微文,欢迎转发转载。
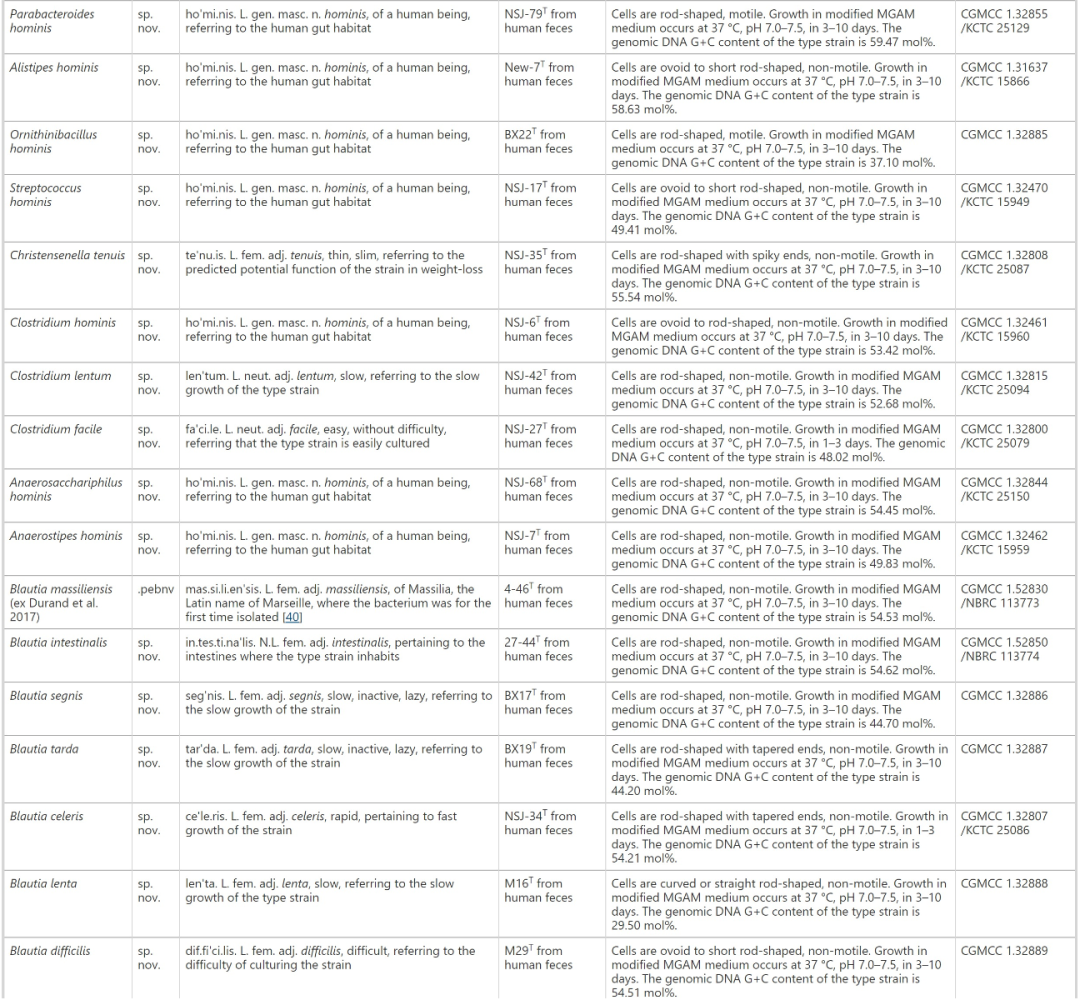
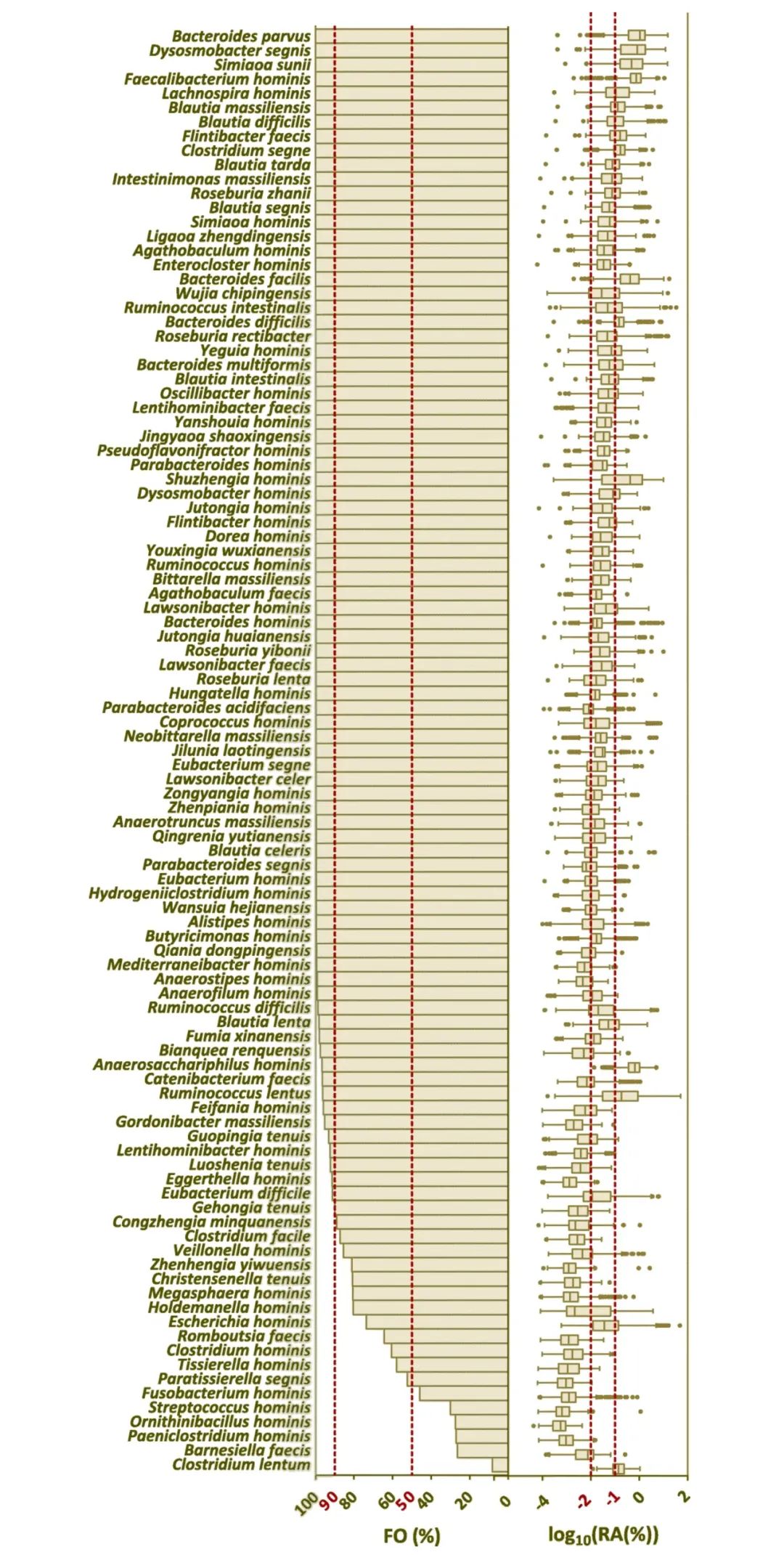
在肠道微生物组学研究中,培养的肠道微生物资源起着至关重要的作用,如帮助揭示肠道微生物的功能和宿主微生物的相互作用。虽然已经进行了几项主要的研究来阐明培养的人类肠道微生物群,但迄今为止,多达70%的人类胃肠道微生物尚未培养。大规模的肠道微生物分离和鉴定以及对公众的可用性是肠道微生物研究和进一步描述人类肠道微生物功能的必要条件。本研究中,作者构建了人肠道微生物生物库(hGMB;主页:hgmb.nmdc.cn),而该研究是通过培养来自中国健康志愿者239份新鲜粪便样本的31份样本混合物中的10558个分离物,并将代表400个不同物种的1170个菌株保存在国际微生物保藏库(中国普通微生物菌种保藏管理中心,CGMCC等)中,以便在全球范围内长期保存和公众获取。根据国际原核生物命名规则,对102个新种进行了特征描述和命名,提出了28个新属和3个新科。hGMB代表了全球人类肠道16S rRNA基因扩增子数据(n=11647)中80%以上的常见和优势人类肠道微生物属和种,并培养了24个“最想要的”和“中等优先”分类群。作者总共测序了115个基因组,代表102个新分类群和13个已知物种。进一步的分析显示,新测序的hGMB基因组代表了统一人类胃肠道基因组(UHGG)中22个以前未培养的物种,并贡献了24个UHGG未发现的潜在“暗物种”。由hGMB基因组产生的非冗余基因目录涵盖了全球最大的人类肠道基因目录中50%以上的功能已知基因(KEGG直系同源),以及FUnkFams数据库中大约10%的“最想要的”功能未知蛋白质。本研究建立了一个可公开获取的人类肠道微生物生物库(hGMB),包含1170个菌株,代表400个人类肠道微生物种类。hGMB通过增加102个新物种、28个新属、3个新科和115个人类肠道微生物基因组,扩展了肠道微生物资源和基因组库,将为推动人肠道微生物研究提供有力的的资源支持。
论文ID
原名:Enlightening the taxonomy darkness of human gut microbiomes with a cultured biobank
译名:用培养微生物库揭开肠道微生物的面纱
期刊:Microbiome
IF:11.607
发表时间:2021.5.21
通讯作者:Shuang-jiang Liu(刘双江)& Chang Liu(刘畅)
通讯作者单位:中国科学院微生物研究所
实验设计

微信视频预览查看
结果










讨论
结论

不感兴趣
看过了
取消

人点赞

人收藏

打赏
不感兴趣
看过了
取消













 010-82736610
010-82736610
 股票代码: 872612
股票代码: 872612
 京公网安备 11010802020745号
京公网安备 11010802020745号










