


















德州大学MD安德森癌症中心发现高选择性的CSF1R抑制剂IACS-9439




安德森癌症中心的研究人员大胆假设,合理设计得出结论。
背景
通过针对PD-1、PDL-1和CTLA4(细胞毒性T淋巴细胞相关抗原4)的检查点抑制剂重新激活先天性和适应性免疫反应来增强抗肿瘤免疫力的策略已显示出良好的临床反应 ,但是,只有一小部分患者表现出持久的临床疗效。临床和临床前证据证明肿瘤相关巨噬细胞(TAMs)作为关键的调节性免疫细胞可以促进肿瘤发展。巨噬细胞主要以两种极化状态存在,交替激活的M2亚型TAM通过分泌抗炎细胞因子(例如IL-10、TGFβ)促进肿瘤发展,而激活的M1亚型TAM通过产生促炎性细胞因子来促进免疫介导的肿瘤杀伤作用。
为了克服TAMs的免疫抑制和促瘤功能,现有治疗策略聚焦于肿瘤微环境中TAM的耗尽及TAM的重编程以推动抗肿瘤功能(致使M2极化到M1)。
集落刺激因子受体(CSF1R)是一个膜相关的受体酪氨酸激酶,是集落刺激因子(CSF1R)的受体。而集落刺激因子可以控制ATMs的产生、增殖和功能。
在一些使用鼠类乳腺癌和成胶质细胞瘤模型的临床前研究中,已证明CSF1-CSF1R信号传导阻滞减慢了原发性肿瘤的生长,减少了肿瘤转移,并改善了荷瘤小鼠的长期生存率。
靶向CSF1R被认为是降低ATM,特别是M2亚型AMT的潜在疗法。
然而,发现缺乏III型激酶活性的选择性CSF1R抑制剂极具挑战性。
CSF1R抑制剂BLZ945在临床前模型中抑制肿瘤生长,降低ATM含量,增加肿瘤间质中CD8+T细胞含量,目前处于临床I//II期,单用或与PD-1单克隆抗体PDR001联用。其抑制CSF1RIC50值为1nM,对其他相关激酶有大于3200倍的选择性。
非选择性的PLX3397是一个非选择性CSF1R抑制剂(抑制靶点包括cKIT、FLT3、PDGFRα和PDFFRβ),已经获得FDA批准上市。
GW2580抑制CSF1RIC50值为30nM,对TrkB和C的选择性分别为22到75倍,对其他相关激酶超过150倍。JTE-952对TrkA的选择性为20倍,而对TrkB和C的选择性未见报道。

优化
研究者基于骨架跃迁策略,使用苯并噻唑作为基本母核,并寻求铰链和连接基的合理组合。
应用MOE中的BREED,在BLZ945结合的CSF1R分子模型中,比较了PDB数据库中1945个预置配体的激酶配体复合物与苯并噻唑的复合物,从而得到了既保持苯并噻唑核心位置又与激酶铰链区相互作用的新型化合物。结果显示,当苯并噻唑与铰链部分之间使用两个原子的连接基,可以使铰链残基Cys666与其形成有效的相互作用。

为了验证该猜测,研究者探究了几个含有两个原子的连接基,结果表明,包含4-吡啶基及-C-O-连接基的化合物3活性最佳。
在吡啶的邻位引入一个氨基,由于其与Cys666的羰基形成相互作用,活性提高6倍,而仅仅在吡啶的3位引入氯,活性也得到提高。当将氨基与氯同时引入,活性提高75倍。

分子对接模型预测,环己基伸入CSF1RDFG-out构象的深口袋,此区域的修饰可能提高活性及选择性(原因在于III型激酶该区域的序列有细微差异)。
为了使小分子与深口袋中Glu633的侧链形成相互作用,在环己基上引入羟基,并分别验证了四个立体异构体的活性,然而,羟基的引入并未显著提高体外活性。
为了评价羟基的引入及羟基的构型是否影响选择性,研究者测试了该系列化合物与一组与CSF1R相关的代表性III型激酶的结合活性,包括FLT3、cKIT、PDGFRα及β。其中化合物12拥有最佳选择性。

化合物12在1μM浓度下,仅仅对468个激酶中的少量III型激酶有明显的结合活性(包括cKIT, FLT3, PDGFRα,和PDGFRcKIT, FLT3, PDGFRα, and PDGFRβ)。

化合物12在临床前种属中显示了合适的体内PK——具有较低的清除率,合理的半衰期及较好的口服生物利用度。

考虑到化合物12的体内外特性,对其进行进一步的体内评估,并与BLZ945进行对比,结果表明,化合物12减少肿瘤巨噬细胞和BLZ945效果基本一致。

然而研究者对化合物12的整体活性、PK性质及选择性水平仍不满意。因此他们将进一步优化其活性、PK及选择性。
为了提高其选择性,分析比较了CSF1R活性位点和cKIT、FLT3、PDGFRα及β的序列及结构并发现CSF1R的Gly795残基与上述激酶的半胱氨酸是并行的。苯并噻唑的4位取代可以占据临近CSF1R-Gly795的空间,同时该位置取代由于空间位阻(与半胱氨酸)在cKIT、FLT3、PDGFRα及β中不耐受。此外,化合物12与高选择性CSF1R抑制剂GW2580的交叠也证明了研究者的假设。

在苯并噻唑4位引入甲氧基后选择性得到极大提高。此外,Thr663和Met637的侧链形成的疏水口袋可以容纳苯并噻唑7位的取代,然而该取代不利于选择性。
在4位引入取代基后选择性得到提高,然而DMPK性质未达到要求,特别是其代谢性质。
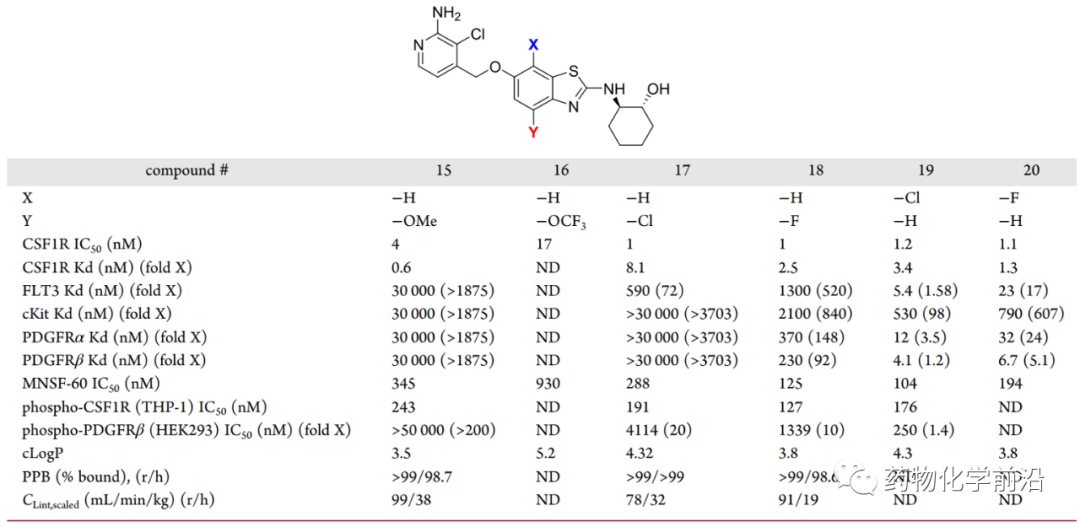
研究者在分析100个类似物的cLogP和肝微粒体清除率的关系后发现,6-甲氧基苯并噻唑系列cLogP大于3.5的化合物倾向于有较高的清除率。基于此,研究者设计并合成了系列cLogP低于3.5的化合物。
通过铰链区的结构修饰降低cLogP提高了代谢稳定性同时保持了选择性。在7位引入氯进一步提高活性的同时并未削弱选择性及PK性质。

尽管化合物22、23显示了杰出的激酶活性、合适的ADME性质,然而细胞活性有待提高。在氨基吡啶氨基上引入乙酰胺后(化合物25),细胞活性得到明显提高。

然而化合物25由于酰胺结构易水解,代谢稳定性并未达到改善。基于此,研究者将易水解的酰胺替换为脲键、吡唑、三唑等取代基,综合评估其激酶细胞活性及代谢稳定性,最终优选出了IACS-9439(1)。

在1μM浓度下,IACS-9439对97个激酶无明显抑制活性(除去CSF1R),对FLT3、cKIT、PDGFRα、PDGFRβ各自有9500倍、>17000倍、1900倍、450倍的选择性。对上述激酶的选择性优于BLZ945。

IACS-9439在小鼠和大鼠中具有合适的体内PK(适中的清除率与半衰期,杰出的口服生物利用度),然而在狗和猴子中,PK性质表现稍差。

在MC38肿瘤模型中,化合物12、IACS-9439、BLZ945均剂量依赖性的减少巨噬细胞数量。在PANC02肿瘤模型中,相同剂量给药IACS-9439比BLZ945,前者比后者具有更低的EMR1 mRNA及CD4 mRNA浓度。
此外,在MC38肿瘤模型中,IACS-9439剂量依赖性的增加M1亚型巨噬细胞含量,减少M2亚型局势细胞,说明该抑制剂可以促进巨噬细胞朝向M1亚型的极化。

总结
肿瘤相关巨噬细胞(TAMs)在多种人类恶性肿瘤的肿瘤间质中均有显著存在,可以促进肿瘤生长。
CSF1-CSF1R信号通路调节ATMs的产生、分化及功能。靶向CSF1R被认为是降低ATM,特别是M2亚型AMT的潜在疗法。此外,在某些癌症中,CSF1R在肿瘤细胞上的高表达与较差的生存率相关,CSF1R是一个潜在的治疗靶点。
然而,发现缺乏III型激酶活性的选择性CSF1R抑制剂极具挑战性。
安德森癌症中心的研究人员大胆假设,合理设计,通过仔细分析对比CSF1R,cKit,FLT3,PDGFRα和PDGFRβ的DGF-out构象中的活性位点,快速发现了高选择性的CSF1R抑制剂IACS-9439,其思路值得借鉴。
参考文献
Barbara Czako, etal. Discovery of IACS-9439, a Potent, Exquisitely Selective, and OrallyBioavailable Inhibitor of CSF1R. J. Med. Chem. 2020. DOI: 10.1021/acs.jmedchem.0c00936


人点赞

人收藏

打赏









 010-82736610
010-82736610
 股票代码: 872612
股票代码: 872612
 京公网安备 11010802020745号
京公网安备 11010802020745号










