
管存在这种总体趋势,我们观察到在少数罕见的大肠杆菌基因中存在强烈选择的证据。
生科云网址:https://www.bioincloud.tech
编译:微科盟Moon,编辑:微科盟居居、江舜尧。
微科盟原创微文,欢迎转发转载,转载须注明来源《微生态》公众号。
导读 微生物基因编码了地球上生命的大部分功能。然而,尽管越来越多研究进行生境宏基因组测序,但目前对全球生物圈的基因分布仍然知之甚少,这对人类和地球的健康都有影响。本研究从14个主要栖息地的13,174个公开宏基因组中构建了一个包含3.03亿个物种水平基因(以95%的核苷酸同源性聚类)的非冗余基因目录,并利用它来表明大多数基因都是特定于单一栖息地的。在多个栖息地发现的一小部分基因富含抗生素抗性基因和可移动遗传元件。通过进一步将这些物种水平基因聚类到3200万个蛋白质家族中,我们观察到小部分蛋白质家族囊括了大部分基因(0.6%的蛋白质家族占50%的基因)。大多数物种水平的基因和蛋白质家族都很罕见。此外,物种水平的基因(特别是稀有基因)显示出较低的正向(适应性)选择率,这支持了一种模型,即在每个蛋白质家族中观察到的大多数遗传变异是中性或接近中性的。
论文ID
原名:Towards the biogeography of prokaryotic genes
译名:原核生物基因生物地理学
期刊:Nature
IF:69.504
发表时间:2021.12
通讯作者:Luis Pedro Coelho,Jaime Huerta-Cepas,Peer Bork
通讯作者单位:复旦大学类脑智能科学与技术研究院,德国欧洲分子生物学实验室
DOI号:10.1038/s41586-021-04233-4
实验设计
结果与讨论
1 全球微生物基因目录 在这项研究中,我们通过整合宏基因组和完整基因组数据,调查各栖息地的原核生物基因,以了解这些基因的全球分布和它们所编码的分子功能。我们整理了来自14个栖息地(包括宿主相关栖息地和环境栖息地;图1)的数据,构建了一个综合的、一致处理的非冗余全球微生物基因目录(GMGCv1)。 GMGCv1来源于13,174个公开的高质量宏基因组(方法,附表1,2)。采用半人工管理的方法对底层样品进行生境标注。对每个宏基因组进行组装和预测ORFs,产生了2,007,736,046个ORFs(方法,附图1,附表3)。为了增加基因目录的覆盖范围,我们纳入了来自proGenomes2数据库的84,029个高质量基因组的312,020,843个ORFs。使用基于图形的冗余去除算法,所得到的2,319,756,889条序列,与之前的生境特定基因目录一样,以95%的核苷酸同源性(大致相当于物种边界的阈值)进行聚类,产生了302,655,267个簇。每个簇中保留一条序列,代表95%核苷酸同源性的所有核苷酸变体,这对应于每个物种的一个特定基因的拷贝,以下称为“单基因(unigene)”。为了能够概括全球基因分布的特征,我们还使用基于同源性的聚类方法对序列进行了更广泛的分组,其依据是统计学意义上的序列相似度和四个额外的氨基酸同一性阈值(>90%、>50%、>30%和>20%)。至少90%同一性的要求是蛋白质数据库中一个严格普遍的阈值,以此产生了210,478,083个独特蛋白质簇,而考虑所有具有统计学意义的、至少20%氨基酸同一性的同源序列,则产生了31,992,232个定义广泛的蛋白质家族。 当前宏基因组学的一个不可避免的限制是,大多数组装的contigs相对于ORFs的长度来说很短,由此产生了许多不完整的ORFs。由于一些分析可能受益于对单个序列质量的严格控制(以较低的覆盖率为代价),并且由于GMGCv1中68.5%的单基因被预测为不完整的ORFs,我们创建了一个只包括完整ORFs的基因目录版本,并且还建立了不同严格程度的可操作定义蛋白家族(https://gmgc.embl.de)。不完整的ORFs和不同的可操作蛋白家族定义都有可能影响功能和系统发育解释。因此,虽然我们在此关注最宽泛的可操作蛋白家族定义(统计学上显著的序列相似性,至少有20%的氨基酸同一性,包括所有的ORFs),但我们所有的观察结果在测试的几个阈值以及包含不完整的ORFs时都是可靠的(附表)。 GMGCv1中的大多数物种水平单基因包含在一小部分大蛋白家族中(0.6%的最大蛋白家族包含了一半的物种水平单基因(图1d))。作为参数定义方面结果的稳健性的一个例子,当完全考虑完整的ORFs(0.5%)或选择更严格的蛋白家族定义时(例如,在50%的聚类阈值时为0.9%;附表4),这部分结果仅有少许变化。因此,在GMGCv1中观察到的遗传多样性主要来源于蛋白质家族内的多样化,而不是基因的从头产生。
接下来,我们尝试将这些基因放到基因组背景中,并产生了278,629个MAGs。即使不去除低质量组装结果(方法,附表5),这些MAGs只包含4000万个物种水平的单基因,而完整目录中的单基因为3.03亿。然而,与以前的结果一致的是,因为MAG优先利用较高丰度的基因,这个MAG子集足以对来自经过充分研究的栖息地的短reads进行映射(在经过充分研究的人类肠道宏基因组中,95.3%的reads映射到MAGs,但42.5%的单基因没有比对到;附图3,4)。
图1 全球微生物基因目录。a,组装来自14个不同栖息地的宏基因组数据,并提取ORFs。这些ORF与来自proGenomes2的ORFs相结合,聚类形成物种水平的单基因、蛋白质簇和蛋白质家族。b,栖息地之间的单基因共享很少,哺乳动物肠道微生物群之间的共享除外。每个条带的宽度代表左边栖息地中共有基因的平均丰度。最宽的带将猫肠道与人类肠道连接起来,代表猫肠道微生物群中58.0%的reads映射到与人类肠道共有的基因。c,单基因积累曲线显示,一些栖息地每个样本的复原率逐渐降低,而其他栖息地(如海洋和土壤)的样本数不足。插图,对于人类肠道,最普遍基因的曲线达到了饱和。然而,罕见的单基因,包括样本特异性基因,仍处于被发现阶段。d,最大的蛋白质家族包含73,979个单基因。然而,从分布模式来看,一半单基因仅包含在203,431(0.6%)个蛋白质家族中(包含≥239个物种水平单基因的家族),而80%的蛋白质家族仅由一个或两个基因组成,仅占总单基因库的不到8%。 2 大多数基因具有生境特异性 虽然MAGs通常是按样品或按生境进行组装的,但全球微生物基因目录使我们能够发现生境之间共有的基因。由于物种水平单基因代表多个序列(核苷酸同源性大于95%),因此它们可能代表了来自多个生境的基因(“多生境基因”)。这些基因可能包含在多个栖息地大量存在的物种中,或者是在栖息地之间流动的物种的一部分,即可以在基因组之间水平转移和跨栖息地边界的基因。只有18,145,135个物种水平单基因(占总数的5.8%)是多生境基因(图1b,附图5)。这与物种倾向于适应其环境,以及在与宿主相关的微生物组中,同种菌株含有宿主特异性基因的发现相一致。为了解开基因跨越栖息地边界的机制(即整个物种或移动元件),我们首先寻找与移动元件相关的单基因,发现它们确实比一般单基因(5.8%)更有可能出现在多个栖息地(1182749个中有156738个(13.3%);附图6)。抗生素抗性基因(ARGs)被认为是频繁携带的移动元件。正如预期的那样,ARGs比其他单基因更有可能出现在多个栖息地(3,208,187个ARG中的329,857个(10.3%);附图6)。为了量化栖息地之间的物种重叠现象,考虑到许多物种尚不为人所知,我们为每个栖息地构建了宏基因组物种(MGSs)作为具有可靠栖息地信息的物种代表。总体而言,我们共构建了7443个MGSs,其中只有1099个MGSs在不同的栖息地之间共享,这与我们观察到的单个单基因的共享模式一致(附图5,cf.图1b)。结果如预期所料,物种更有可能在相似的环境中共享(附图7);例如,不同的哺乳动物肠道栖息地共享许多MGSs(1099个中有786个共享)。 3 丰富度模式具有生境特异性 为了调查每个样本中同种基因的存在,我们使用普遍存在的单拷贝基因的丰富度来衡量分类丰富度,并将其与整体单基因的丰富度进行比较。我们观察到每个样本中每个物种的物种水单基因的平均数量明显不同(图2a)。海洋和土壤环境表现出多种子模式的混合。就海洋样本而言,这些子模式对应不同的海洋深度,特别是将浅水样本与那些在无法接触到阳光照射的深水中采集的样本进行比较的情况下,而土壤环境中的差异则与酸度和湿度不同有关(附图8)。因此,每个物种在宏基因组中存在的单基因数量可以成为一个界定良好的栖息地的识别特征。 为了检验观察到的单基因丰富度主要由包含多个同源单基因(假定具有相同的代谢功能)或多种功能组的群落驱动,我们计算了蛋白质家族丰富度与物种水平单基因丰富度的比值,作为功能冗余的表示方式,并观察到栖息地之间的明显差异(图2b)。我们进一步测试了生境特异性,仅利用4个描述符(分类、系统发育、单基因和蛋白家族丰富度)构建了预测每个样本生境的分类器。通过交叉验证,我们评估这一分类方法的准确率为86.1%。已在多种环境中描述了功能冗余,即多种生物编码相同的功能。尽管它否定了每个代谢生态位由单一物种占据的简单模型,但对于解释它的过程或其含义仍没有达成共识。从我们的数据中,我们得出结论,每个环境中的功能冗余与群落发展的栖息地密切相关,这与对泛基因组的分析结果一致。因此,功能冗余的一般模型将需要纳入特定的栖息地参数。
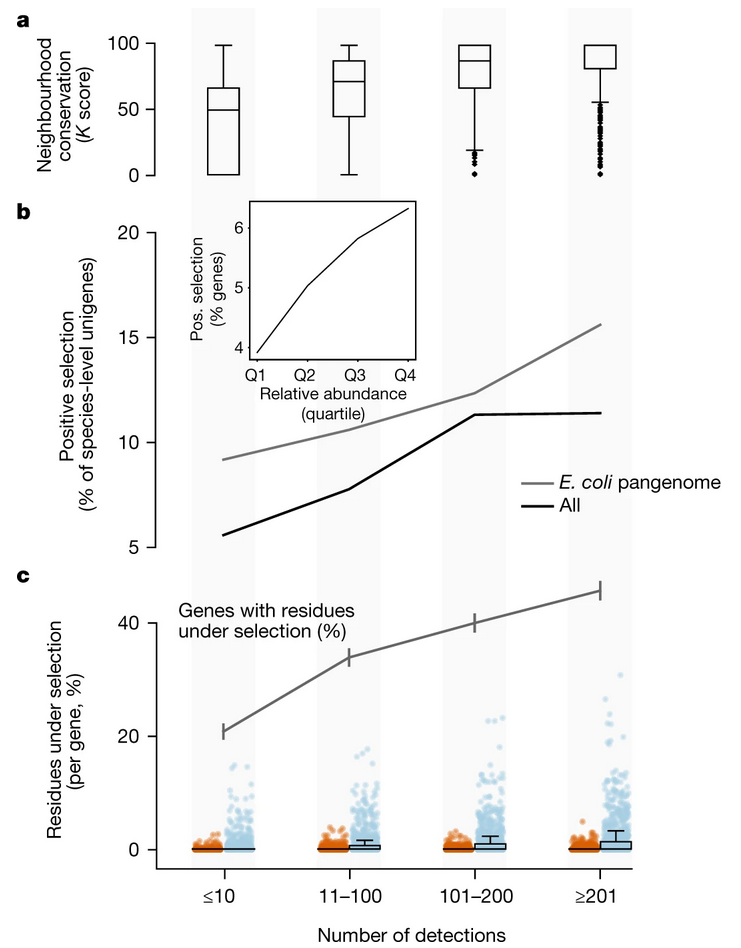
图2 在每个宏基因组中,同种基因的数量和功能冗余表明在生境内的差异明显小于生境间的差异。a,按栖息地划分的每个样本中同种基因数量的密度,表明每个样本中最大的泛基因组存在于环境样本中,而不是与宿主相关的栖息地。b,每个栖息地、每个样本中检测到的每个蛋白家族的单基因数量密度(功能冗余的代表)在不同栖息地之间有明显差异。在研究充分的人类肠道栖息地,蛋白质家族丰富度与使用eggnog-mapper2获得的更严格的同源丰富度估值高度相关,并扩展到所有栖息地。 4 大部分基因很罕见 在确定了功能冗余和大多数基因的生境特异性后,我们分析了宏基因组中单基因的频率。我们观察到,物种水平单基因的流行率呈指数规律,每个栖息地的范围不同(图3),这清楚地表明大多数基因的流行率很低。事实上,如果我们把在10个或更少的样本中检测到的基因视为罕见基因,那么GMGCv1中的大多数单基因是罕见的(54.7%的基因,附图9,附表4)。在中性(或接近中性)进化的假设下,这些指数规律形式的频率分布是符合预测的,并且很好地解释了我们的数据(对于人类肠道,单基因的理论拟合和观察数据之间的Pearson相关性为0.997,附表6)。 与该模型一致,绝大多数蛋白质家族由罕见、低丰度的簇组成,围绕着物种水平的单基因,没有进一步的同源基因(图1d,附图10)。正如在完全测序的基因组中观察到的那样,没有可检测到的同源物的基因预计对生物体的适应性几乎没有(如果有的话)影响,并且在环境背景下也应如此。由于操纵子(operon)结构,功能可以通过相邻基因的共现来推断,因此,我们测量了跨流行类别的基因顺序和通路邻域的保守性。稀有物种水平的单基因在功能上的相互作用确实比流行基因的少(图4a),这与稀有基因受到的进化限制较少相一致。 然后,我们通过分析序列变异来研究我们的数据是否符合中性进化模型。中性意味着大多数观察到的遗传差异对适应性没有影响,因此不是由于对特定生态位的适应(正向选择),尽管净化(负向)选择可能仍然活跃。由于蛋白家族之间的选择操作不同,我们在每个蛋白家族中测试了正向(适应性)选择。我们发现,绝大多数的单基因并没有显示出正向选择的证据(图4b)。然而,我们观察到罕见单基因比常见单基因(高达10%)适应性要小得多(4%)(图4b)。为了防止物种间进化速度和流行程度的差异以及可能存在的技术问题可能产生的混淆效应,我们只使用了GMGCv1中5126个注释良好的大肠杆菌基因组中的单基因,得到了非常相似的正选择增加和基因流行程度的相关性(图4b)。此外,GMGCv1中现有的大肠杆菌基因组的数量足以测试每个位点的选择压力,事实上,这表明罕见的大肠杆菌单基因位点比那些更常见的位点所承受的选择压力要小(图4c)。然而,在单个基因组中,大多数基因既不处于低选择压力下,也不罕见。在5126个大肠杆菌基因组中,每个基因组中只有2.8%±1.7%的基因是罕见的(也就是说,它们出现在我们收集的10个或更少的宏基因组中)。然而,不同栖息地的大肠杆菌菌株库十分巨大,与此相对应的是,大肠杆菌的泛基因组,像大多数其他细菌一样,是开放的,因此其基因组将共同包含大量的稀有基因。 由于我们的采样和测序深度对非常稀有的基因存在偏倚,虽然我们无法量化生态和进化过程对观察到的模式的相对贡献,也无法证明稀有基因近乎中性进化的现象,但我们观察到的相关性指向这样一种模式,并表明我们可能仍然低估了稀有基因的数量。因此,随着测序成本的不断降低,我们也许能够捕获地球上所有丰富的原核生物物种,因为这一目标似乎已经在人类肠道等经过充分研究的栖息地实现了。从我们的数据来看,对于生物多样性非常高的栖息地(如土壤)似乎也是可行的。然而,由于大量稀有的、生境特异性的、甚至可能是特定于地区的基因,以及可能发生的从头产生、修饰和灭绝的周转过程,全球基因库的相当一部分基因可能永远不会被捕获。
图3 大部分基因很罕见。正如中性或近中性进化模型所预测的那样,基因流行率的直方图在对数尺度上大致呈线性。
图4 罕见的单基因受到较低的选择压力。a,操纵子结构在普遍存在的基因中被保留的概率更大。b,在可检测到的正向选择下单基因的比例随着检测次数的增加而增加。这在大肠杆菌泛基因组中也成立。c,大肠杆菌泛基因组是唯一有足够规模来测试每个位点选择压力的基因组。大肠杆菌泛基因组中的高丰度基因显示出比每个位点的罕见基因更强的反向(蓝色)和正向(红色)选择的证据。尽管存在这种总体趋势,我们观察到在少数罕见的大肠杆菌基因中存在强烈选择的证据。例如,我们发现有利于抗生素抗性的UDP-葡萄糖6-脱氢酶基因的例子,尽管只在6个样本中观察到选择的证据。箱线图显示了中位数和四分位数,须延伸到最远的数据点。 原文链接: https://www.nature.com/articles/s41586-021-04233-4 获取此篇微文原文pdf请扫描下方二维码联系微科盟多组学老师即可。
微文推荐阅读
土壤微生态文献包免费领取 | 150篇近两年高影响因子土壤微生态相关文章
根际微生态文献包免费领取 | 60篇近两年高影响因子植物根际微生物相关文章
肠道微生态文献包免费领取 | 100篇近两年高影响因子肠道菌群与消化道疾病相关文章
元素循环微生态文献包免费领取 | 60篇近两年高影响因子植物-土壤碳氮磷相关文章
获取此文献原文PDF、申请加入学术群,联系您所添加的任一微科盟组学老师即可,如未添加过微科盟组学老师,请联系组学老师45,无需重复添加。
了解更多菌群知识,请关注“微生态”。
点击阅读原文,直达原文网站,如需原文PDF可联系组学老师获取
不感兴趣
看过了
取消

人点赞

人收藏

打赏
不感兴趣
看过了
取消
©2012-2023 北京华媒康讯信息技术股份有限公司 All Rights Reserved. 注册地址:北京 联系电话:010-82736610
广播电视节目制作经营许可证 —(京)字第 17437号 京海食药监械经营备20200522号
京ICP备12011723号 京ICP证150092号
 京公网安备 11010802020745号
工商备案公示信息
互联网药品信息服务资格证书((京)-非经营性-2020-0015)
京公网安备 11010802020745号
工商备案公示信息
互联网药品信息服务资格证书((京)-非经营性-2020-0015)






您已认证成功,可享专属会员优惠,买1年送3个月!
开通会员,资料、课程、直播、报告等海量内容免费看!

打赏金额
认可我就打赏我~
1元 5元 10元 20元 50元 其它
打赏作者
认可我就打赏我~
扫描二维码
立即打赏给Ta吧!
温馨提示:仅支持微信支付!
已收到您的咨询诉求 我们会尽快联系您




































 010-82736610
010-82736610
 股票代码: 872612
股票代码: 872612




