


















科研 | mSphere:中东呼吸综合征 (MERS) 冠状病毒与重症MERS患者微生物的扩增子和宏基因组分析




中东呼吸综合征 (MERS) 冠状病毒与重症MERS患者微生物的扩增子和宏基因组分析
编译:微科盟流浪过的梦,编辑:微科盟茗溪、江舜尧。
导读
中东呼吸综合征冠状病毒(MERS-CoV)是2012年在中东出现的一种人畜共患传染病。症状从轻微到严重不等,包括呼吸道和胃肠道疾病。该病毒主要存在于骆驼种群中,偶尔会传染给人类。人类感染的严重程度受多种因素的影响,类似于严重急性呼吸系统综合症冠状病毒2(SARS-CoV-2),潜在的健康并发症可能起主要作用。目前,MERS-CoV和SARS-CoV-2在中东同时发生,需要一种快速的方法对MERS-CoV进行测序,以获得分子流行病学的基因型信息。此外,中东呼吸综合征冠状病毒感染的复杂因素是跟临床相关的合并感染。快速表征这些感染的能力是有利的现象。为了迅速对MERS-CoV进行测序,我们开发了一种基于扩增子的方法与纳米孔阵列的RNA直接测序相结合。这种方法提供了关于共有基因组和微小变异存在的数据,包括缺失突变体。而宏基因组分析提供了背景微生物组的信息。这种方法的优点是可以识别插入和缺失。这是冠状病毒基因型变化的主要驱动因素。
论文ID
原名:Amplicon and Metagenomic Analysis of Middle East Respiratory Syndrome(MERS)Coronavirus and the Microbiome in Patients with Severe MERS
译名:中东呼吸综合征(MERS)冠状病毒与重症MERS患者微生物的扩增子和宏基因组分析
期刊:mSphere
IF:4.389
发表时间:2021.7.21
通讯作者:Waleed Aljabr
通讯作者单位:沙特阿拉伯法赫德国王医疗城研究中心&英国新发和人畜共患病卫生保护研究组
DOI号:10.1128/mSphere.00219-21
实验设计

背景
冠状病毒曾被描述为病毒学的死水,因为它们不会在人类中引起广泛的疾病。然而,随着2003年中国出现严重急性呼吸综合征冠状病毒(SARS-CoV)、2012年沙特阿拉伯出现中东呼吸综合征冠状病毒(MERS-CoV)以及现在2019年起源于中国的SARS-CoV-2,情况发生转变。这些病毒会引起呼吸道和胃肠道疾病,并具有相似的基因组结构和疾病谱。患者现有的医疗问题,例如心血管疾病和遗传因素,可以表现出更严重的症状和潜在地具有致命的结果。因此,其他呼吸道病原体的存在在这些感染的病因中或许是至关重要的,它们也可能是正常健康微生物组的一部分。人类严重感染的典型特征是细胞因子风暴、肺炎和肾衰竭。中东呼吸综合征冠状病毒已被世卫组织确定为优先疾病,并在其紧急情况下用于研究和开发的病原体列表上。随着SARS-CoV-2的出现,迫切需要制定通用的医疗对策来对抗这些感染并减轻未来的爆发,尤其是在接触者跟踪和了解传播动态方面,迫切需要未满足的医疗保健需求。结果,这些病毒削弱了国家基础设施,引发了全市正常交通并扰乱了国际旅行和商业。许多国家同时面临SARS-CoV-2和MERS-CoV的风险。
所有这三种病毒的出现之间存在惊人的相似之处。例如,中东呼吸综合征冠状病毒的主要问题之一是朝觐期间病毒和其他病原体的潜在传播。这是一年一度的沙特阿拉伯麦加伊斯兰朝圣之旅,涉及全球约200万人口和约50万沙特居民。从骆驼到人类的传播事件不断发生,通过传播和/加强监测,单峰驼中MERS-CoV的地理分布正在增加。目前,尚无抗病毒疗法或疫苗被批准用于治疗或预防人类冠状病毒感染。多项研究正在评估针对MERS冠状病毒感染的医疗对策。这些包括已经在临床中使用的化合物,例如干扰素-α2b和利巴韦林。由美国陆军部资助的基于病毒刺突糖蛋白的1期DNA疫苗最近在一项人体试验和单峰骆驼进行了评估。此外,已在沙特阿拉伯开始了一项人体1期临床试验,以评估基于ChAdOx1 MERS疫苗的安全性和耐受性。零星爆发和缺乏合适的动物模型阻碍了该项研究。
由于缺乏医疗控制措施,切断传播对于控制此类爆发至关重要,而对病毒基因组进行测序可以通过分子流行病学协助,正如新加坡的SARS-CoV所证明的那样。测序提供有关病毒进化速度、特定聚集性病源以及疫情是否由持续的人际传播引起的信息;或者它可以用来调查人畜共患病爆发。以牛津纳米孔平台为中心的测序方法使用,迅速减少了测序周转时间,可以应用于冠状病毒,例如SARS-CoV-2。
MERS-CoV具有高度传染性,沙特阿拉伯王国及周边地区正在陆续爆发。应用基于扩增子和宏基因组MinION测序方法,为了通过快速生成病毒基因组序列和绘制鼻咽样本中的微生物组来帮助诊断MERS-CoV。在冠状病毒复制过程中,利用大约30 kb的正义RNA基因组,合成了一组嵌套的亚基因组mRNA。位于基因组3,端的亚基因组mRNA通常比靠近5,端的转录本更丰富,这些产物可以通过扩增子测序进行鉴定。本研究联合使用扩增子测序和宏基因组方法研究来自MERS患者的临床样本。扩增子方法基于生成跨越基因组但取决于起始引物的30、15或8个重叠片段。使用更大的扩增子的基本原理是建立一个系统,该系统可用于识别基因组中的缺失或重组位点,这是冠状病毒RNA合成的标志。数据表明,中东呼吸综合征冠状病毒的全基因组或近全基因组信息是从感染患者的样本中快速获得的,并使用扩增子方法进行测序。此外,基于扩增子的测序方法提供了关于共有基因组和微小变异存在的数据,包括缺失突变体。宏基因组分析提供了背景微生物组的信息以及这如何与结果相关联。对患者微生物组的实时分析和抗生素抗性标志物的识别可能会提供更好的化疗方法来管控感染。
结果与讨论
单核苷酸多态性、重组和由此产生的缺失(以及潜在的插入)可能是在某些冠状病毒株中观察到的广泛基因组多样性的原因。这些重组可能导致新的冠状病毒株或可能影响疫苗策略。为了通过MERS-CoV测序来识别这些基因组变化,开发了牛津纳米孔提供的快速读取方法。他们使用了通过位于MERS-CoV基因组上的适当引物对选择的更长的扩增子(图1)。请注意,此方法还将使用嵌套的亚基因组mRNA集来捕获序列信息。

图1 保守引物对(表4)在MERS-CoV基因组上的位置及与MERS-CoV基因相比的位置。引物对可用于生成不同长度的扩增子,包括所示的30、15和8个扩增子。MERS-CoV基因组上显示了适当的基因。
表4 本研究纳入患者的特征

1 使用从MERS-CoV感染细胞中纯化的总RNA的引物验证和扩增产物的生成
为了评估所选引物在受控条件下扩增病毒RNA的效用,从感染复数(MOI)为5的MERS-CoV EMC株感染的MRC-5细胞中纯化总RNA。该RNA用作模板以使用随机六聚体启动cDNA合成。扩增条件见表1;MERS-CoV基因组使用30-扩增子(图2A)、15-扩增子(图2B)或8-扩增子(图2C)进行扩增,对每组扩增子使用相同的一组条件(见表S1在补充材料中)。在所有引物对中使用相同扩增条件背后的基本原理是,如果需要大规模样本分析,扩增效率会更高。这些数据表明,对于所有方法,都观察到跨越MERS-CoV基因组的PCR产物。然而,15-和8-扩增子方法产品的效率各不相同。
表1 30-、15-和8-扩增方法的PCR条件。


图2 使用30(A)、15(B)和8(C)种引物对组合产生的扩增子的琼脂糖凝胶电泳。这些引物对用于与从MERS-CoV感染细胞中提取的RNA的逆转录相结合生成扩增子,并在凝胶图像的适当泳道上方显示。分子大小标记显示在左侧。
2 从感染MERS-CoV的患者中产生扩增子并检测分析出共有基因组序列
30-、15-和8-扩增子方法在从MERS患者的鼻腔抽吸物中提取的RNA上进行了评估。数据表明30-和15-扩增子方法可用于从临床样本中生成片段(图3A和B)。8-扩增子方法不足以从临床样本中获得整个MERS-CoV的覆盖率(数据未显示),可能是因为RNA的质量低于从细胞培养中获得的RNA质量。将30-和15-扩增子方法(来自不同患者)中产生的PCR产物对每位患者进行组合、质控,并对每位患者进行单独测序。MinION生成的测序读数与参考序列对齐。分析表明,从30-扩增子(图4A)和15-扩增子方法(图4B)中可以获得完整的基因组序列。使用ARTIC生物信息学分析生成了一个共有序列。为了识别占主导地位的病毒基因组序列,使用自定义perl脚本根据参考序列(GenBank登录号NC_019843.3)计算每个核苷酸的数量。该序列用于为个体患者生成定制的共有基因组。

图3 使用30(A)和15(B)引物对组合产生的扩增子的琼脂糖凝胶电泳。这些引物对与从MERS患者的鼻吸液中提取的RNA的逆转录相结合,用于产生扩增子。引物对显示在凝胶图像上的适当泳道上方。分子大小标记显示在左侧。

图4 使用Oxford Nanopore MinION设备在单个流动池上测序的30(A)和15(B)个扩增子的读取深度分析。y轴表示MERS-CoV基因组上每个位置的覆盖范围。MERS-CoV基因组的核苷酸长度由x轴上的基因组位置表示。虚线代表20 x覆盖,表明在这条线以上每个核苷酸至少测序了20次。
3 分析患者中的次要变异群体
感染中的微小变异群体已被证明影响病毒复制的动力学,并与患者的预后相关。因此,开发了一些方法,可用于评估来自中东呼吸综合征冠状病毒感染患者的样本中的微小变异频率。自定义的perl脚本用于调用共识,还可揭示每个位置的核苷酸深度和核苷酸的数量(图5)。深度用于将突变频率标准化为一个比例而不是原始计数,从而可以比较不同读取深度的样本。在分析中会去除计数小于20的核苷酸。作为依据,这种方法被应用于从患者10和115获得的测序数据,因为它们的质量更高。患者10似乎比患者115具有更多的碱基变化(图6)。突变(A > G、G > A、C > U 和 U > C)更常见,并且C > U似乎比其他突变更突出。这一发现与 APOBEC的RNA编辑一致。APOBEC 3家族成员已被证明参与限制人类冠状病毒NL 63(HCoV-NL 63)的生长,并被确定为SARS-CoV-2中C到U转换的潜在驱动因素。我们注意到,使用MinION测序数据评估次要变异群体可能存在问题,因为与基于Illumina的方法相比,测序错误率更高。然而,在样本中有足够的读取深度时,仍然可以获得次要变异频率/总体的情况。通过将每个MERS基因除以总病毒基因组的长度(30,108 bp)来计算调整后的基因大小。全局indel多态性按类别分组,并将其映射到参考序列中的相应位置(图7)。每个观察的平均比例(相对于深度)乘以调整后的基因大小,得到每个基因的标准化插入/缺失比例(图8)。

图5 是用于导出病毒基因组覆盖度和患者体内病毒基因组的微小变异信息的生物信息学的分析流程图。
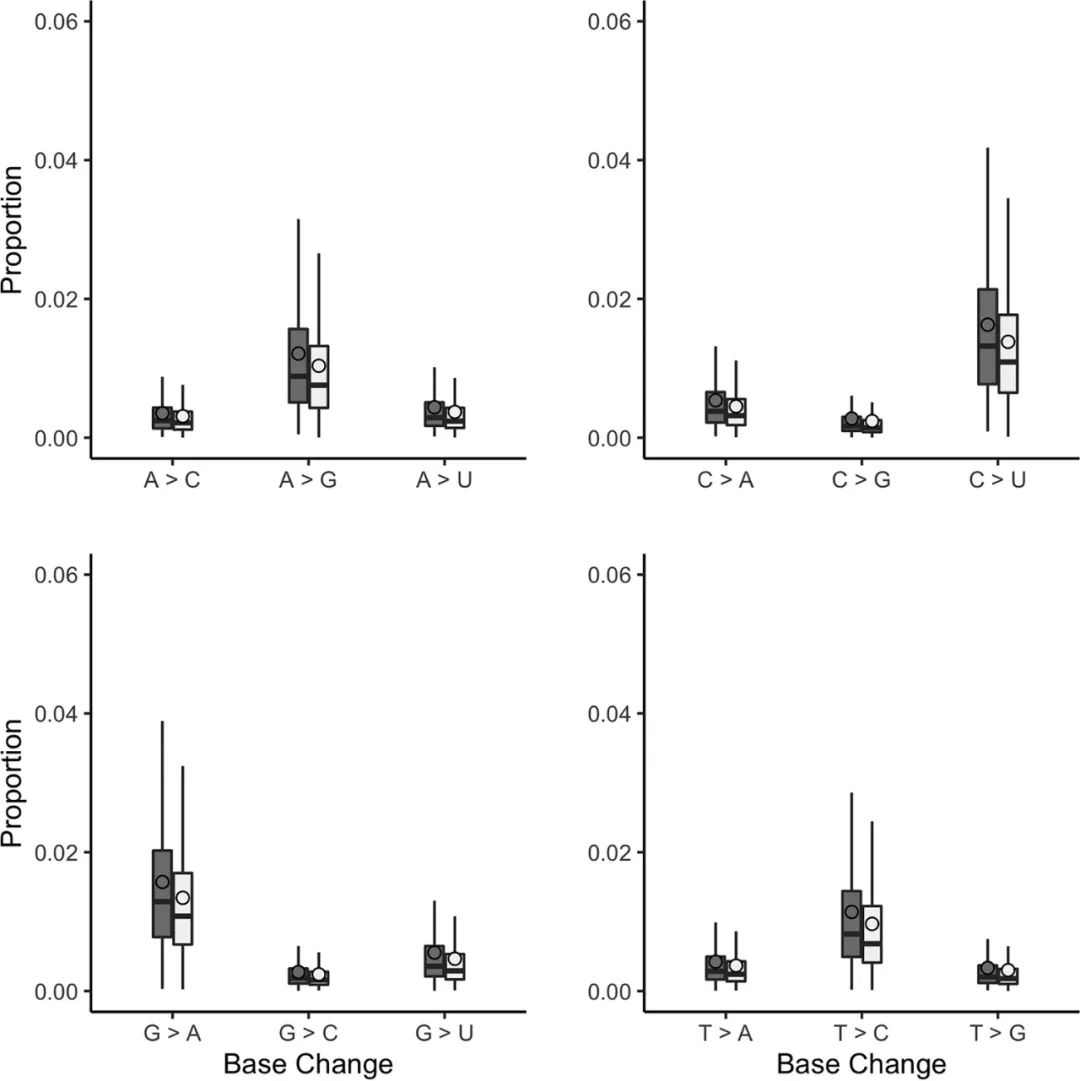
图6 测序读数被映射到患者共有病毒基因组序列。自定义脚本计算每个基因组位置的每个碱基的数量,质量得分 >10。从分析中删除深度< 20的位置。该图显示了与患者显性共有参考基因组相比观察到的碱基变化比例。总体而言,观察到的转变比颠换更频繁,其中 C > U 是观察到的最多的碱基变化。患者10,深灰色;患者115,浅灰色;未可视化的异常值。

图7 患者(PX)110和PX 115的标准化插入缺失多态性的全基因组表示。插入缺失由条形表示并锚定在x轴,对应于每个基因。标准化的indel比例在每个图中排名。在规模差异阻碍可视化的情况下,径向子图用于提供具有最低插入缺失比例的基因的视图。

图8 indel多态性的全基因组表示,由PX 110和PX 115的平滑密度(调整,0.05;α,0.7)证明。
4 患者样本中病毒基因组缺失的鉴定和分析
在来自患者10和115的测序数据中发现了MERS-CoV基因组中的缺失(表2)。患者10在Orf1ab中有五个缺失,一个跨越N的缺失。患者115使用15个扩增子方法进行测序,因此生成长度超过2 kb的扩增子。在该患者中发现了跨越orf4b和orf5基因的77个碱基的缺失。该区域的缺失可能对病毒的发病机制有影响,ORF4A能够抑制宿主的早期抗病毒反应(干扰素α/β [IFN α/β]),同样ORF5可以减少炎症反应。这一发现在使用的两个生物信息学系统中都是一致的。这些缺失的存在并不排除在次要变异水平上存在完整的基因。然而,数据确实说明了MERS-CoV基因组的可塑性。
表2 患者115和10的MERS-CoV基因组中存在的缺失分析。

5 鉴定患者样本中的细菌和病毒序列以及抗生素抗性标记
MERS患者中其他感染的存在也可能需要临床管理并影响结果。宏基因组学方法用于鉴定MERS患者临床样本中存在的其他微生物。鉴定出鲍曼不动杆菌(Acinetobacter baumannii)、铜绿假单胞菌(Pseudomonas aeruginosa)、肺炎链球菌(Streptococcus pneumoniae)等物种,均与细菌性肺炎有关(图9)。尽管RNA的检测并不总是推断活动性感染,但识别是基于活动的细菌转录组。

图9 来自15名严重MERS感染患 者的前20种属。 从测序文库中删除人类读数,并使用 Kraken2鉴定病毒和细菌转录本。 Kraken2输出在使用Phyloseq导入R之前被转换为biom 格式。 为每个患者绘制了每个物种的相对丰度。 +,检测到MERS-CoV读数; −,未检测到 MERS-CoV读数。
具有不动杆菌(Acinetobacter)转录物的患者与该疾病致命结果相关(图9)。不动杆菌感染通常只出现在医疗机构中,尤其是接受吸氧的患者中。不动杆菌属被认为是一种严重的多重耐药病原体,包含在ESKAPE的缩写中,指的是屎肠球菌(Enterococcus faecium)、金黄色葡萄球菌(Staphylococcus aureus)、肺炎克雷伯菌(Klebsiella pneumoniae)、鲍曼不动杆菌(Acinetobacter baumannii)、铜绿假单胞菌(Pseudomonas aeruginosa)和肠杆菌属(Enterobacter species)。在肺炎克雷伯菌(TEM-4)和鲍曼不动杆菌(LpxC、adeI、adeJ、mexT、adeN、adeK和ADC-2)内的患者6中鉴定了抗生素抗性基因的证据(表3)。该患者死于严重的MERS感染。几个团体建议,广泛使用抗生素作为COVID-19患者管理的一部分可能会影响抗菌素耐药性(AMR)。如本研究所示,对来自严重冠状病毒感染患者样本的宏基因组分析可用于追踪这些标记。通过这种方法在患者5中鉴定了人类alphaherpesvirus 1转录本。在整个SARS-CoV-2大流行期间正在进行的研究表明,冠状病毒或许能够在严重疾病期间重新激活单纯疱疹病毒和巨细胞病毒。
表3 中东呼吸综合征冠状病毒感染患者细菌中发现的耐药基因。

使用DESeq 2比较致命病例和非致命病例之间的细菌丰度(图10)。一般来说,变形杆菌门细菌的转录本在致死病例中的丰度高于非致死病例,包括不动杆菌属和克雷伯氏菌属的物种。以前的研究表明病毒感染和细菌群落之间存在相互作用。在慢性阻塞性肺疾病(COPD)患者中观察到实验性鼻病毒感染后变形菌的丰度增加。发现2009年甲型H1N1流感感染和肺炎患者的变形菌与非病毒性肺炎的比较。
数据表明,基于扩增子的方法可用于对临床样本中的MERS-CoV进行快速测序,并提供有关遗传多样性和插入/缺失的信息。这种方法与能够解析临床样本中存在的微生物组的宏基因组方法相辅相成。该分析确定了与通风相关的细菌以及抗生素抗性标志物。总体而言,该研究证明了快速直接测序在表征来自 MERS人类样本中的MERS-CoV感染特征方面的效用。

图10 与源自转录本丰度数据的非致死病例相比,MERS-CoV感染致死病例中细菌丰度的增加和减少。将Phyloseq对象转换为DESeq2对象,使用致命和非致命结果之间的对比来计算log 2倍变化,并绘制错误发现率(FDR)<0.01的值。x轴表示已识别的转录本的属,未分配指的是不能在属水平被分配的转录本,颜色表示门。

不感兴趣
看过了
取消

人点赞

人收藏

打赏
不感兴趣
看过了
取消













 010-82736610
010-82736610
 股票代码: 872612
股票代码: 872612
 京公网安备 11010802020745号
京公网安备 11010802020745号










